Article Text

Abstract
Inclusions of pathogenic deposits containing TAR DNA-binding protein 43 (TDP-43) are evident in the brain and spinal cord of patients that present across a spectrum of neurodegenerative diseases. For instance, the majority of patients with sporadic amyotrophic lateral sclerosis (up to 97%) and a substantial proportion of patients with frontotemporal lobar degeneration (~45%) exhibit TDP-43 positive neuronal inclusions, suggesting a role for this protein in disease pathogenesis. In addition, TDP-43 inclusions are evident in familial ALS phenotypes linked to multiple gene mutations including the TDP-43 gene coding (TARDBP) and unrelated genes (eg, C9orf72). While TDP-43 is an essential RNA/DNA binding protein critical for RNA-related metabolism, determining the pathophysiological mechanisms through which TDP-43 mediates neurodegeneration appears complex, and unravelling these molecular processes seems critical for the development of effective therapies. This review highlights the key physiological functions of the TDP-43 protein, while considering an expanding spectrum of neurodegenerative diseases associated with pathogenic TDP-43 deposition, and dissecting key molecular pathways through which TDP-43 may mediate neurodegeneration.
- ALS
- motor neuron disease
- motor physiology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
TAR DNA-binding protein-43 (TDP-43), initially identified in 1995 as a suppressor of HIV-1 (HIV-1) gene expression, is a highly conserved and ubiquitously expressed RNA/DNA-binding protein belonging to the heterogeneous nuclear ribonucleoprotein (hnRNP) family.1 TDP-43 is pivotal in multiple cellular functions including regulation of RNA metabolism, mRNA transport, microRNA maturation and stress granule formation.1 In line with its nuclear and cytoplasmic functions, TDP-43 can shuttle between the nucleus and the cytoplasm, but under normal physiological conditions, localisation is predominantly nuclear.2 Of relevance to brain function, TDP-43 appears to be critical for normal development of central neuronal cells in early stages of embryogenesis.3 Given the extensive role of TDP-43 in cellular processes, particularly in the development of the central nervous system, it is unsurprising that the dysfunction of TDP-43-related pathways has been increasingly recognised as an important pathogenic mechanism in neurodegenerative disease.
In 2006, hyperphosphorylated and ubiquitinated TDP-43 cytoplasmic inclusions were identified as a pathological feature of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar disease (FTLD).4 5 Pathogenic missense mutations in the TARDBP gene, which encodes the TDP-43 protein, were subsequently identified as causative genetic mutations in both ALS and FTLD, although in a small percentage of familial cases. Interestingly, the vast majority of patients with ALS and FTLD do not harbour mutations in the TARDBP gene yet demonstrate widespread abnormalities involving TDP-43. The pathophysiological heterogeneity of ALS and FTLD phenotypes may suggest that multiple pathogenic pathways contribute to mislocalisation and aggregation of TDP-43. Over the past decade, TDP-43 deposition has been associated with an increasing number of neurodegenerative diseases, where it has been identified as the primary pathogenic factor, resulting in these disorders being designated as ‘TDP-43 proteinopathies’.6
The following review examines the neurodegenerative disorders associated with TDP-43, relating these disorders to structure and function of TDP-43, while also discussing the clinical phenotypes and potential pathophysiological mechanisms. Finally, the potential utility of therapeutic agents that target the pathophysiological processes linked to TDP-43 proteinopathies will be appraised.
Structure and function of TDP-43
TDP-43 is a conserved hnRNP containing 414 amino acids and encoded by the TARDBP gene (1.p36.22).7 The protein structure is comprised of an N-terminal region, nuclear localisation signal (NLS), two RNA recognition motifs: RRM1 and RRM2, nuclear export signal (NES), a C-terminal region encompassing a prion-like glutamine/asparagine-rich (Q/N) domain and a glycine-rich region1 8 9 (figure 1A). Although predominantly localised in the nucleus, TDP-43 shuttles between the nucleus and the cytoplasm, a process mediated by active and passive transport,2 where it exerts physiological functions (figure 1B). In addition, TDP-43 localises to the mitochondria where it associates with the mitochondrial genome and is important in the respiratory chain pathways.10

(A) Structure of TAR DNA-binding protein 43 (TDP-43) protein. The TDP-43 protein contains 414 amino acids and is comprised of an N-terminal region with a nuclear localisation signal (NLS). In addition, the protein consists of two RNA recognition motifs (RRM1 and RRM2), a nuclear export signal (NES) and a C-terminal domain with a glutamine/asparagine-rich (Q/N) and glycine-rich regions. Mitochondrial localisation motifs (M1; M3; M5) are also evident. Pathogenic mutations are predominantly located within the C-terminal region which can exhibit prion-like properties. The numbers represent amino acid lengths. (B) The TDP-43 protein is critical for mediating RNA metabolism. In the nucleus, TDP-43 is important for transcription and splicing of messenger RNA (mRNA), as well as maintaining RNA stability (pA) and transport to nucleus. In addition, TDP-43 regulates biogenesis of microRNA (miRNA) and processing of long non-coding RNA (lncRNA). Although predominantly located within the nucleus, TDP-43 shuttles between the nucleus and the cytoplasm. In the cytoplasm, TDP-43 participates in mRNA stability, translation, formation of stress and ribonucleoprotein (RNP) transport granules.
The N-terminal domain is important in the formation of functional homodimers, which are critical for proper TDP-43 physiological function.1 Located within the N-terminal region is the NLS domain which mediates the import of TDP-43 into the nucleus where it exerts its physiological functions.1 The RNA-binding motifs (RRM1 and RRM2) are essential for TDP-43 protein binding to RNA/DNA molecules, regulating transcription, translation, splicing and stability of mRNA, as well as mediating RNA export.11 In addition, TDP-43 forms ribonucleoprotein (RNP) granules which are important for the transportation of mRNA molecules and for promoting biogenesis of non-coding RNAs, such as microRNA (miRNA).12 13 Separately, the prion-like C-terminal glycine-rich region has been implicated in TDP-43 pathogenesis as this region regulates protein solubility and mediates pathological aggregation.14 TDP-43 is also important for the formation of stress granules, protecting the neuronal cells against cellular insults such as oxidative stress.15 Given that TDP-43 exerts important functions in neuronal cells, it could be reasonably anticipated that mutations and dysfunction of TDP-43 lead to severe neurological diseases.
Genetics of TDP-43 proteinopathies
As discussed below, TDP-43 proteinopathies encompass a wide range of neurodegenerative diseases and phenotypes, which may be inherited in a Mendelian pattern or be apparently sporadic. A large number of genes have been associated with TDP-43 proteinopathies (table 1). Interestingly, genetic abnormalities are associated with multiple phenotypes and diseases. Specifically, the C9orf72 hexanucleotide expansion may cause ALS, FTLD, ALS-FTLD, Alzheimer’s disease (AD) phenotypes and atypical Parkinsonism. The mechanisms underlying these pleiotropic effects are unclear, although genetic and/or environmental factors impacting on gene expression have been proposed but remain to be identified. The notion of ALS being a multistep disease,16 with fewer steps required in familial ALS (fALS),17 underscores the importance of epigenetic and environmental factors in ALS pathogenesis.
Diseases associated with TDP-43 pathology
Although several TDP-43 proteinopathy genes encode RNA-binding proteins, the functions of the other associated genes are diverse. From this, we can conclude that TDP-43 pathology may arise through multiple different mechanisms. Identifying the relationship between these dysfunctional genetic and molecular processes may be critical for development of effective therapies.
Pathogenesis of TDP-43
The precise pathophysiological mechanisms by which TDP-43 causes neurodegeneration remain to be elucidated. Experimental evidence suggests that biological dysfunction in multiple cellular processes may contribute to neuronal death (figure 2).1 The observation of the histopathological hallmarks of TDP-43 proteinopathies, specifically cytosolic mislocalisation and increased nuclear clearance of TDP-43, has led to the suggestion of two non-mutually exclusive disease mechanisms, namely, nuclear loss-of-function and cytoplasmic gain-of-function. Underscoring this notion are mouse model studies disclosing that overexpression of TDP-43 is associated with neurotoxicity, while ‘knocking-out’ the TARDBP gene is embryonically lethal.18 A limitation of most mouse TDP-43 models pertains to the fact that TDP-43 protein levels, which are physiologically narrowly autoregulated, are significantly increased or suppressed.19 Thus, the toxicity observed in these models, which can occur with overexpression of mutant or wild-type protein or conditional deletion, may not be representative of the underlying pathobiology.

Putative mechanisms by which TAR DNA-binding protein 43 (TDP-43) exerts its pathogenic effects in TDP-43 proteinopathies. Multiple processes have been implicated in TDP-43 pathogenesis. Nuclear depletion of TDP-43 results in dysregulation of RNA metabolism resulting in reduced protein production. Cytoplasmic aggregation of hyperphosphorylated TDP-43 (depicted by blue P) is a hallmark of TDP-43 proteinopathies and may result in cellular stress, aberrant stress granule formation, mitochondrial dysfunction, reduced autophagy and dysfunction of proteosomal processes. In addition, inhibition of endocytosis and reduced expression of heat shock proteins have also been reported, as well as prion-like behaviour (cell-to-cell spread). In addition to hyperphosphorylation, other post-translational modifications of the TDP-43 protein include cysteine oxidation (depicted by green C), which leads to pathogenic oligomerisation, as well as ubiquitination (leading to increased aggregation), acetylation (resulting in impaired RNA binding, mitochondrial dysfunction and aggregation), and PARylation (promotion of phase separation of TDP-43 into stress granules).
TARDBP gene mutation
Pathogenic missense mutations in the TARDBP gene (>50 mutations) have been reported in both sporadic and familial cases of ALS and FTLD,20 with the majority of mutations clustering in the C-terminal glycine-rich region which enhance the aggregation propensity and cytotoxicity of the TDP-43 protein, thereby leading to a toxic gain-of-function. The C-terminal region also interacts with the other hnRNPs to mediate RNA splicing, thereby providing an additional avenue for pathogenesis. Mutations in the putative amyloidogenic core region of the TARDBP gene renders the TDP-43 protein vulnerable to formation of amyloid-like fibrils, which are characteristics of ALS/FTLD inclusions, with subsequent neuronal toxicity.4 In addition, TARDBP mutations may induce cytoplasmic mislocalisation of TDP-43 leading to further neuronal toxicity. The TDP-43 protein is also intricately associated with stress granule formation, and TDP-43 mutations result in larger stress granules, with reduced distribution density and mobility, thereby impairing TDP-43-mediated RNA homeostasis and leading to neuronal dysfunction.1
Dysregulation of RNA metabolism
Support for dysregulation of RNA metabolism as a pathogenic mechanism in TDP-43 proteinopathies has been provided by mouse model studies.21 22 Specifically, an ethylnitrosourea generated C-terminal TARDBP mouse mutant with isogenic levels of expression has been shown to lead to late onset motor neuron degeneration preceded by genome-wide RNA changes.22 Also a low copy transgenic model revealed that disease-associated mutant TARDBP, but not wild-type TARDBP, leads to progressive weakness which is associated with deficits in stress granule assembly.21 In addition, knock-in mice approaches have also demonstrated that patient-derived TDP-43 mutations lead to perturbed RNA metabolism and pathogenesis.23 24 Intranuclear localisation of TDP-43 has also been reported to exert an important role in RNA metabolism. Specifically, TDP-43 was shown to associate with Gemini of Cajal bodies (gems), which are important in gene expression.25 In ALS, aberrant accumulation of TDP-43 leads to a loss or decrease of gems, leading to defective spliceosome function and ultimately neurodegeneration.26 27
Nuclear TDP-43 depletion
A prominent pathological feature of all TDP-43 proteinopathies is nuclear depletion of the TDP-43 protein, which is mostly seen in the end stages of the disease process.1 As TDP-43 shuttles between nucleus and cytoplasm, it engages in diverse functions within both compartments (figure 1B). In the nucleus, TDP-43 regulates many aspects of RNA metabolism, including mRNA splicing, and a nuclear loss-of-TDP43 leads to degradation and reduction of RNAs as well as altered splicing events.28 29 Transcriptional dysregulation and splicing defects are therefore likely to contribute to neuronal dysfunction in TDP-43 proteinopathies. While multiple TDP-43 reliant RNA targets are likely to be associated with neuronal dysfunction, stathmin-2 has emerged as an attractive candidate, with reduced expression of this protein reported in transdifferentiated neurons from ALS patient-derived fibroblasts expressing TDP-43 mutations.30 Interestingly, reduced stathmin-2 expression was associated with poor axonal regenerative capacity, which was restored by normalisation of stathmin-2 protein expression.
Disordered nuclear transport
Nuclear localisation signal and NES regulate the nucleocytoplasmic shuttle of TDP-43, and deletion of these segments leads to depletion of the TDP-43 nuclear pool, thereby impacting on the regulation of chromatin assembly and histone processing resulting in nuclear aggregation.1 Of relevance, aggregation of mutant TDP-43 was shown to trigger sequestration and mislocalisation of nucleoporins and other transport factors, thereby interfering with nucleocytoplasmic transport of proteins and RNA.31 Nuclear pore pathology has been identified in the brain tissue of patients with sporadic and familial (TARDBP) ALS. Interestingly, mutations in the NLS region have been infrequently reported in ALS, although mutations in the C-terminal region may also drive cytoplasmic localisation.32 In the cytoplasm, TDP-43 aggregation may serve as a repressor of translation through interactions with ribosomal proteins.10 Compounds modulating nucleocytoplasmic transport warrant further investigation as these may be of therapeutic potential.
Post-translational modification and pathogenic oligomers
Post-translational modifications of the TDP-43 protein may also contribute to neurotoxicity in TDP-43 proteinopathies (figure 2).1 Phosphorylation of TDP-43 is a pathological feature of ALS and FTLD,4 evident in both the cortex and the spinal cord. Multiple phosphorylation sites were identified in the carboxyl-terminal regions of deposited TDP-43, and the TDP-43 phosphorylation led to increased oligomerisation, fibrillisation, cytoplasmic mislocalisation and aggregation in neuronal cells, thereby contributing to pathogenesis (figure 3).32 33 Ubiquitination, acetylation, poly ADP-ribosylation and cystein oxidation are additional post-translational modifications that can induce TDP-43 aggregation and impair its ability to interact with the DNA/RNA and protein–protein interactions.1

{kind=link}
{kind=link}
{kind=link}
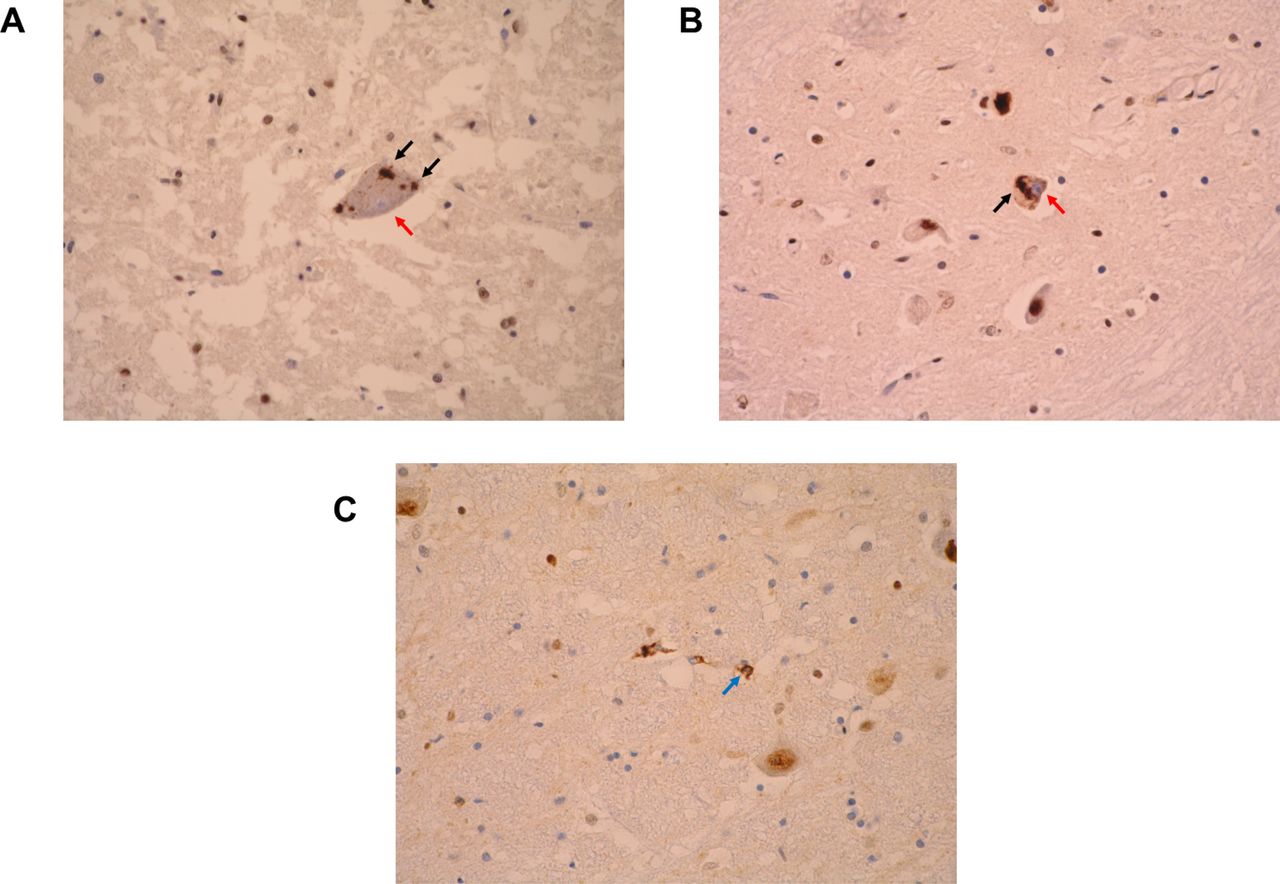
Neuropathological hallmarks of TDP-43 proteinopathies as observed with TDP-43 immunohistochemistry. (A) An alpha motor neuron in the cervical spinal cord of a patient with amyotrophic lateral sclerosis (ALS) demonstrating intraneuronal cytoplasmic inclusions of TDP-43 (black arrows) and nuclear clearance (nucleus stained blue, red arrow). (B) Intraneuronal cytoplasmic aggregates (black arrow) and nuclear clearance (nucleus stained blue, red arrow) in a neuron of the inferior olivary nucleus. (C) Glial coiled body-like inclusions (blue arrow) in the caudal pons. All images represent light microscopy micrographs obtained at ×400 magnification (University of Newcastle). The antibody used was the proteintech antibody (Proteintech, rabbit polyclonal; product code 1072-2-AP). All images are derived from patients with sporadic ALS.
Pathogenic TDP-43 oligomers have also been reported in ALS and FTLD, which are structurally distinct from physiological oligomers, and possess an increased propensity to cytoplasmic aggregation and neurotoxicity.34 In addition, the pathogenic TDP-43 oligomers are capable of cross-seeding with Alzheimer’s amyloid-β protein and exhibit prion-like behaviour. The notion of centrifugal propagation of TDP-43 pathology, whereby pathogenic TDP-43 aggregation begins in the prefrontal cortex and propagates along axonal fibres,35 may explain the prion-like spread observed in ALS. A similar pathological observation has been reported in FTLD.36 Taken together, the prion-like activity of the pathogenic TDP-43 protein may explain the rapid and relentless clinical evolution of TDP-43 proteinopathies.
Liquid–liquid phase separation
Of further relevance, formation of membraneless spherical liquid droplet-like organelles by proteins containing prion-like domains, through a process called liquid–liquid phase separation (LLPS), is an increasingly recognised pathophysiological mechanism in neurodegenerative diseases.1 Under physiologically stressful conditions, TDP-43 may be recruited to stress granules and this may be an intermediate step between soluble and aggregated TDP-43. By means of its unstructured, aggregation-promoting prion-like C-terminal domain, TDP-43 can rapidly separate from the aqueous solution and enter gel-like membraneless structures, such as stress and RNA granules. Although LLPS is entirely reversible in wild-type TDP-43, mutations in the C-terminal region may disrupt the dynamics of formation and disassembly of these granules resulting in protein aggregation (figure 3).37 Phase transfer of cytoplasmic TDP-43 may also occur independently of stress granule formation. Ultimately, abnormalities of the LLPS process result in inhibition of nucleocytoplasmic transport, increased clearance of nuclear TDP-43 and neuronal degeneration. Interestingly, post-translational modification of TDP-43, by addition of a poly(ADP-ribose) polymer to its NLS sequence, was shown to promote LLPS and protein aggregation.38 Importantly, downregulation of tankyrase, a poly(ADP-ribose) [PAR] polymerase, prevented LLPS and neurodegeneration, suggesting a potential therapeutic utility of tankyrase modulation in TDP-43 proteinopathies.
Autophagy
Additionally, TDP-43 is an important regulator of autophagy, critical for the clearance of pathogenic TDP-43 oligomers.39 Inclusion bodies positive for autophagy markers have been identified in some TDP-43 proteinopathies such as ALS and FTLD, and have been associated with disease progression.40 Interestingly, mutations in the ALS-associated ubiquilin-2 (UBQLN2) gene have been shown to impair autophagy, inducing an overall increase in TDP-43 and promoting TDP-43 aggregation in neuronal cells.41
Mitochondrial dysfunction
Mitochondrial dysfunction also appears to be an important mechanism of TDP-43 toxicity (figure 2).1 9 Specifically, overexpression of pathogenic TDP-43 in motor neurons leads to reduction of mitochondrial length, impairment of mitochondrial movement, distribution and dynamics, such as fission and fragmentation.10 In addition, mitochondrial dysfunction has been shown to exacerbate TDP-43 toxicity, which, in turn, amplifies the degree of oxidative stress. Intriguingly, the interaction between TDP-43 aggregation and oxidative stress appears to amplify the cellular TDP-43 toxicity as well as the deleterious effects of TDP-43 on mitochondrial function.1 Importantly, blocking the mitochondrial localisation of TDP-43 has been shown to mitigate its cellular toxicity and to improve the clinical phenotype in animal models.10 Recently, TDP-43 and its disease-associated mutants have been found to significantly enhance mitochondrial abnormalities across various models, thereby reflecting the mitochondrial dysfunction observed in patients with ALS.10 Consequently, the interaction between the pathogenic TDP-43 and mitochondria appears to be a crucial mechanism of eliciting toxicity in TDP-43 proteinopathies.
Additional neurotoxic mechanisms have also been implicated in TDP-43 proteinopathies and include impairment of endocytosis, dysregulation of metal ion homeostasis and interference with chromatin remodelling.1 The importance of these processes in the pathogenesis of TDP-43 proteinopathies requires further clarification.
In addition to TDP-43 accumulation in the central nervous system, increased TDP-43 protein levels accompanied by a reduction in TDP-43 mRNA levels in peripheral blood mononuclear cells (PBMCs) has been reported in patients with sporadic ALS.42 Importantly, the aberrant expression of TDP-43 resulted in dysfunction of critical molecular processes, such as downregulation of molecular chaperone HSC70, responsible for recognition, binding and translocation of protein substrates into lysosomes. Additionally, cytoplasmic localisation of TDP-43 has also been reported in human skin fibroblasts, resulting in specific molecular defects.43 When combined with novel biomarkers, including assessment of disrupted cortical networks,44 and measurement of neurofilament light chain,45 as well as adoption of time-to-event trial endpoints,46 the use of PBMCs and skin fibroblasts may provide unique research opportunities, especially in drug screening strategies.
Cytoplasmic aggregation of TDP-43 has also been demonstrated in glial cells,47 48 predominantly oligodendrocytes, although the pathological implications remain to be clarified. While TDP-43 appears to be important for proper functioning of mature oligodendrocytes, depletion of TDP-43 leads to necroptosis of mature oligodendrocytes and downregulation of myelin proteins, but not lower motor neuron degeneration.49
Diseases associated with TDP-43 inclusions ‘TDP-43 proteinopathies’
Amyotrophic lateral sclerosis
The pathophysiological mechanisms underlying ALS are likely to be multifactorial and mediated by a complex interaction between dysfunction of vital molecular pathways and genetic mutations.16 50 51 Abnormalities of critical molecular cellular processes such as glutamate excitotoxicity, mitochondrial dysfunction, oxidative and endoplasmic reticulum stress together with ion channel dysfunction and genetic mutations are all likely to contribute to ALS pathogenesis. Multiple environmental risk factors have been identified, including smoking, repeated head injury and physical activity, but exert small effects.52 Importantly, cortical hyperexcitability has been identified as an important pathophysiological mechanism in ALS,53 preceding the onset of lower motor neuron dysfunction,54 mediating disease progression55 and underlying the development of clinical features of ALS such as dissociated muscle atrophy.56 57
Of the genetic mutations linked to ALS, increased hexanucleotide repeat expansions in C9orf72 (40%) gene are the most frequent, followed by missense mutations in SOD1 (20%), FUS (1%–5%) and TARDBP (1%–5%) genes.7 Importantly, 40%–60% of the sporadic (sALS) risk is thought to be genetic, with repeat expansions in several genes (ATXN1, ATXN2 and NIPA1) and multiple single nucleotide polymorphisms being associated with sALS. In addition, 10% of patients with sALS carry pathogenic genetic mutations, mostly C9orf72, thereby suggesting that any differences between familial and sALS might be arbitrary.
The varied clinical manifestations have led to the reclassification of ALS as a primary brain neurodegenerative disorder.58 59 Underscoring this notion have been neuropathological findings of frontal and temporal lobar atrophy associated with neuronal loss, superficial linear spongiosis and ubiquitinated tau-negative and synuclein-negative intraneuronal inclusions that contain hyperphosphorylated TDP-43 proteins.1 4 Strikingly, TDP-43 aggregation is evident in ~97% of all ALS cases.47 These TDP-43 inclusions are evident in both demented and non-demented patients with ALS, and increase in density with disease evolution particularly the development of cognitive impairment.60 In ALS, three predominating cell-type specific patterns of TDP-43 pathology have been identified, including (1) glial (22% of cases); (2) mixed neuronal and glial (59% of cases); (3) neuronal (7% of cases).61 The extent of TDP-43 pathology differentiates ALS-FTLD from ALS without FTLD,62 and the presence of TDP-43 pathology in extra-motor areas was associated with cognitive impairment in ALS, as can be assessed by the Edinburgh Cognitive and Behavioural ALS Screen (ECAS).63 TDP-43 pathology in the orbitofrontal, dorsolateral prefrontal, medial prefrontal cortices and ventral anterior cingulate were associated with executive dysfunction. Language dysfunction was associated with TDP-43 pathology in the inferior frontal gyrus, transverse temporal area, middle and inferior temporal gyri, as well as the angular gyrus. Verbal fluency dysfunction was associated with TDP-43 pathology in the prefrontal cortex, inferior frontal gyrus, ventral anterior cingulate and transverse temporal area. Behavioural abnormalities, however, were associated with TDP-43 pathology in the orbitofrontal and prefrontal cortices as well as the ventral anterior cingulate.63
Other motor neuron diseases
Primary lateral sclerosis (PLS) and progressive muscular atrophy (PMA) represent atypical variants of motor neuron disease, clinically characterised by either isolated upper motor neuron or lower motor neuron dysfunction, respectively.64 65 In the 2015 revision of the El Escorial criteria, PLS and PMA were considered forms of ALS,66 being labelled as restricted phenotypes with slower disease progression and better prognosis, a notion that has been supported in the recent revision of the ALS diagnostic criteria.59 Cognitive and behavioural dysfunction, within the spectrum of FTLD, has been documented in PLS and PMA, suggesting that neurodegeneration is not limited to the motor system and challenging the concept of restricted phenotypes.67 A limited number of neuropathological studies have reported TDP-43 inclusions in both PLS and PMA phenotypes.68 In patients with PLS, phosphorylated TDP-43 positively stained neuronal cytoplasmic inclusions (NCIs) and dystrophic neurites/neuropil threads were evident in the frontotemporal cortex and subcortical grey matter, while these pathological changes were scarce in the spinal motor neurons.68 In PMA, three different pathological patterns have been reported, with the most frequent being widespread pathological TDP-43 inclusions in the upper and lower motor neurons, including neocortical and limbic areas, resembling ALS.69 The remaining two patterns include LMN degeneration with TDP-43 pathology without UMN degeneration, and a combination of UMN and LMN degeneration with FUS-positive basophilic inclusion body disease.69 Although more research is needed on PLS and PMA, clinical, pathological and genetic evidence suggest that these disorders are TDP-43 proteinopathies and potentially part of the FTLD-ALS continuum.
Frontotemporal lobar degeneration
Three different types of neuropathological changes have been reported in FTLD, including (1) FTLD-tau which accounts for 35%–50% of cases and is associated with specific phenotypes such as Pick’s disease, corticobasal degeneration and progressive supranuclear palsy.70 Tau pathology is associated with mutations in the MAPT gene (17q21.31), which encodes the protein tau, and accounts for 5%–20% of all familial FTLD cases; (2) FTLD-FUS is associated with the behavioural variant (bvFTLD) and manifests as severe disinhibition with psychiatric features. Motor or language deficits are not a prominent feature of FTLD-FUS, which represents approximately 10% of all FTLD cases and is not associated with FUS gene mutations or ALS; (3) FTLD-TDP-43, which accounts for ~50% of the FTLD cases,47 is subdivided into three major neuropathological subtypes based on the patterns of cytoplasmic or intranuclear pathology and cortical association.70 In type A pathology, small compact or crescentic neuronal cytoplasmic inclusions are evident in neurons with short, thin neuropil threads. Clinically, type A pathology appears to be associated with non-fluent primary progressive aphasia (PPA), corticobasal degeneration and bvFTD (with or without ALS). Type B pathology is characterised by diffuse or granular NCIs (with relatively little neuropil threads) and is commonly identified in FTLD-ALS and bvFTLD. Importantly, 15% of patients with FTLD develop ALS, while signs of lower motor neuron dysfunction may be evident in 30%–40% of patients, comparable to the frequency of cognitive and behavioural changes in ALS.70 The genetics of FTLD-TDP-43 overlap with ALS, with C9orf72 hexanucleotide expansions accounting for 25%–40% of the familial FTLD cases.70 In addition, mutations in GRN, C9orf72, VCP, CHMP2b and TARDBP genes are also associated with FTLD-TDP-43 (table 1).71 Type C pathology is characterised by long, tortuous dystrophic neurites which strongly associate with semantic-variant PPA or temporal-variant bvFTD.70
Facial onset sensory and motor neuronopathy
Facial onset sensory and motor neuronopathy (FOSMN) is a rare neurodegenerative disease of motor and sensory neurons, characterised by the onset of sensory symptoms in the trigeminal nerve distribution with slow evolution from face to neck, upper trunk, upper extremities and finally lower extremities.72 Progressive bulbar motor symptoms develop in later stages of the disease, followed by lower motor neuron symptoms in the limbs evolving in a rostral-caudal pattern. Patients with FOSMN develop disabling weakness, bulbar symptoms (requiring gastronomy) and eventually fatal respiratory failure. The disease course therefore is reminiscent of ALS, although progression is slower with an average survival of approximately 8 years.72 To date, 100 cases of FOSMN syndrome have been reported. Heterozygous mutations in several fALS genes have been reported in FOSMN syndrome, including TARDBP, SOD1, SQSTM1, VCP and CHCHD10, although the pathogenic significance of these remains to be determined.73 Although the aetiology of FOSMN syndrome remains to be fully elucidated, the identification of TDP-43 positive intraneuronal inclusions suggests that FOSMN syndrome is a neurodegenerative disorder.74–76 Importantly, some studies have failed to detect TDP-43 inclusions,72 77 suggesting a heterogeneity of the disease process in FOSMN syndrome.
TDP-43 pathology in older adults with cognitive impairment (LATE and CARTS)
Although TDP-43 pathology may be evident in older adults without cognitive changes,78 it may also be observed in neurons of cognitively impaired patients that are distinct to AD.79 Specifically, TDP-43 inclusions may be evident in a condition referred to as hippocampal sclerosis (HS) dementia that may occur in up to 20% of the ‘oldest of old (≥85 years of age)’. Clinically, the dementia presents with episodic memory impairment thereby resembling AD. At a pathological level, marked neuron loss and gliosis in the hippocampal formation is evident, which is out of proportion to AD neuropathological changes, with TDP-43 inclusions evident within the hippocampus.80 81 Two novel entities have been recently described on the basis of TDP-43 distribution and absence or low levels of AD pathology. Limbic-predominant age-related TDP-43 encephalopathy (LATE) refers to a dementia syndrome with absent AD pathology and TDP-43 inclusions (often with coexisting HS) predominantly confined to the limbic system.82 Clinically, LATE is characterised by an amnestic dementia syndrome similar to AD in older adults (25%–50% older than 80). Genetic variation in five genes is associated with LATE, namely, GRN, TMEM106B, ABCC9, KCNMB2 and APOE. Autopsy results of community-based cohorts show that approximately 25% of people with cognitive impairment had neuropathological changes compatible with LATE.82
In addition, cerebral age-related TDP-43 with sclerosis (CARTS) is a neuropathological entity similar to LATE. CARTS should be considered in patients with cognitive impairment in the oldest-of-old (≥85 years of age). TDP-43 pathology is evident in both the hippocampus and the extra-hippocampal regions with low levels of AD pathological changes (Braak NFT stages≤IV) and absence of clinical features of FTLD.80 81
Guam Parkinson-dementia complex (G-PDC) and ALS (G-ALS)
There are a few isolated areas in the world, such as Guam (USA), the Kii peninsula (Japan) and Irian Jaya (Indonesia), where ALS is highly prevalent. Approximately 200 per 100 000 population per year of the indigenous Chamorro people of Guam used to be affected by a progressive cognitive impairment with extrapyramidal signs (G-PDC, bodig) or motor neuron dysfunction (G-ALS lytico). Consumption of cycad nuts contained in flour and boiled fruit bats was linked to both diseases, and attributed to the neurotoxins glucosides and β-methylamino-L-alanine. Since the complete removal of the cycads from the diet, the incidence of G-PDC and G-ALS was significantly reduced (22 per 100 000 population/year), although not completely abolished, suggesting the importance of other causative genetic or environmental factors.83 Although initial neuropathological studies in G-PDC disclosed the deposition of the tau proteins within the neurofibrillary tangles, recent studies have identified TDP-43 positive inclusions in the brain and spinal cord of patients with G-PDC and G-ALS, suggesting that this too is a TDP-43 proteinopathy.84 TDP-43 pathology has also been reported in Kii ALS/PDC from the Hohara focus.85
Multisystem proteinopathy
Multisystem proteinopathy (MSP), also referred to as inclusion body myopathy (IBM) associated with early-onset Paget disease of the bone and FTLD dementia, is a complex autosomal dominant inherited disorder. The clinical phenotype is characterised by a combination of the following clinical features: depression and apathy (most common), weight loss, Parkinsonism and respiratory failure (hypoventilation).86 Inclusion body myositis leads to progressive proximal weakness, developing around the age of 40, and leading to wheelchair dependency within 10–15 years post symptom onset. At a pathological level, TDP-43 and ubiquitin protein inclusions are evident in rimmed vacuoles of muscle fibres.86 The cytoplasmic TDP-43 aggregation was associated with abnormalities of RNA metabolism, suggesting a novel pathogenic basis for IBM.87 Multiple genes have been implicated in MSP, including VCP, MATR3, SQSTM1 and hnRNPA1/B2. These genes harbour RNA binding motifs, similar to the FUS and TARDBP genes, and are critical in regulating the RNA lifecycle such as transcription, splicing, RNA trafficking and sequestration.88
Perry disease
Perry disease is a rare neurodegenerative hereditary disorder, with autosomal dominant inheritance, and caused by mutations in the dynactin-1 gene (DCTN1, 2p13.1).89 The clinical phenotype is characterised by a combination of the following clinical features: depression and apathy (most common), weight loss, Parkinsonism and respiratory failure (hypoventilation).89 Interestingly, DCTN1 mutations may also lead to a fALS and distal hereditary motor neuropathy 7B phenotype. Over 20 families have been identified worldwide, accounting for approximately 100 patients. The mean age of onset is around 50 years (range 35–70 years). The mean disease duration is 5.5 years (range 2–14 years), and the most common cause of death is respiratory failure.89 In Perry disease, TDP-43 pathology is selectively seen in the extrapyramidal system and brain stem, with the distribution being distinct to that seen in ALS or FTLD, being evident in NCIs, dystrophic neurites, perivascular astrocytic inclusions and spheroids.90 Recently, coexisting tau and α-synuclein pathologies have also been identified in Perry disease, suggesting neuropathological heterogeneity.
Disorders with concomitant TDP-43 pathology
(1) Alzheimer’s disease (AD) is the most frequent dementia in adults over the age of 65, presenting with loss of episodic memory, followed by impairment in other cognitive domains and behavioural changes. Pathological hallmarks of AD include neuritic plaques (extracellular deposits of β-amyloid) and neurofibrillary tangles (intraneuronal deposits of insoluble hyperphosphorylated tau protein). These hallmarks are predominantly found in the hippocampal and temporal regions, with emerging evidence that TDP-43 aggregates are evident in up to 57% of AD brains.91
(2) Chronic traumatic encephalopathy (CTE) is a neurodegenerative disorder with unique neuropathological findings caused by repetitive moderately severe traumatic brain injury (TBI). Interestingly, repeated head injury has also been reported to be associated with ALS. It is typically seen in athletes that practised contact sports and military veterans. The clinical presentation of CTE is variable and includes a combination of progressive cognitive, behavioural and mood changes, as well as ALS-like motor deficits.92 Pathological features of CTE include cortical atrophy, predominantly affecting the frontal and temporal cortical regions, cavum septum and ventricular enlargement. Pathologically, CTE is characterised by perivascular accumulations of phosphorylated tau (p-tau) in neurons and astrocytes along with accumulation of β-amyloid, α-synuclein and TDP-43. It is noteworthy that TDP-43 pathology is found in nearly all CTE cases and localises with tau.92
Therapeutic strategies in TDP-43 proteinopathies
Therapeutic strategies aimed at clearing TDP-43 protein aggregations as well as modulating other TDP-43 pathogenic processes may be of utility in TDP-43 proteinopathies.1 93 Small-molecule inhibitors of TDP-43 aggregation were shown to decrease TDP-43 aggregation and prevent cellular toxicity.94 In addition, small-molecule inhibitors were shown to inhibit the accumulation of TDP-43 into stress granules, reduce C-terminal fragment aggregation and enhance caspase-mediated cleavage.95
Increased expression of heat shock proteins (HSPs), which can reduce protein aggregation, may also prove therapeutically useful in TDP-43 proteinopathies. Notably, increased expression of specific HSPs (Hsp104, Sis1, DNAJB6, Hsp40) leads to dissolution of TDP-43 aggregates, reduced in toxicity, suppression of further TDP-43 aggregation and promotion of nuclear localisation,96 with improvement in growth and survival of cells. Heat shock transcription factors (HSFs), important for cellular proteostasis, were shown to reduce the levels of insoluble TDP-43 protein in cell culture and ALS mouse models. Interestingly, the HSF-mediated TDP-43 clearance was closely associated with expression of HSPs (Hsp70, DNAJB2a) which had the potential to further suppress TDP-43 aggregation.97
Nuclear import receptors (NIRs), which are part of the nuclear pore machinery, exert chaperone and disaggregase functions.98 The NIR, karyopherin-β1, decreases and reverses TDP-43 fibrillisation possibly by associating with its nuclear localisation signal sequence. Separately, NIR agents have been shown to reverse TDP-43 fibrillisation and prevent stress granule formation by associating with the NLS sequence, thereby garnering interest for nuclear importins as promising therapeutic targets.99 At this stage, the therapeutic utility of all these compounds targeting TDP-43 clearance and aggregation remains to be confirmed, although considerable interest exists in the small molecules, HSPs, HSFs and nuclear importins as promising therapeutic targets.
Several genetic therapeutic approaches could be used in TDP-43 proteinopathies, including antisense oligonucleotides, interference of RNA pathways through siRNAs, gene delivery techniques and antibody-mediated reduction of protein aggregation.20 While a detailed discussion of the methodologies is beyond the scope of this review (see Chio and colleagues for a more detailed discussion),20 these approaches could provide viable therapeutic options, although are still in the early stages of development.
Given that the autophagy pathways also play an important role in the clearance of misfolded and aggregated proteins, enhancing the autophagy pathways could be of therapeutic importance in TDP-43 proteinopathies. Induction of autophagy by small molecules such as rapamycin enhances protein degradation, improves memory, rescues motor dysfunction and reduces cytoplasmic inclusions in a TDP-43 mouse model.100 The clinical efficacy of autophagy-enhancing agents is currently being assessed in both ALS and Guam ALS/PDC.20
Conclusions
The TDP-43 protein has emerged as a vital protein for maintaining cellular health, by virtue of its manifold functions in RNA metabolism and homeostasis. Underscoring its importance in human disease are findings that aberrations in TDP-43 homeostasis are associated with lethal and progressive neurodegenerative disorders. While the neurodegenerative spectrum of TDP-43 proteinopathies initially included ALS and FTLD, the clinical continuum has significantly expanded, suggesting a central role of TDP-43 in neurodegenerative diseases. TDP-43 appears to exert neurotoxicity by a variety interdependent pathogenic processes, although the mechanisms by which neurodegeneration ensues remains to be fully elucidated. The uncovering of these processes could yield novel therapeutic targets common to a multitude of neurodegenerative diseases.
References
Footnotes
Twitter @jnnp_bmj
MAvE and SV contributed equally.
Correction notice This article has been corrected since it appeared Online First. Author name Hugo M De Oliveira was updated.
Contributors EMJdB, MS, HO, SV, VKO, LvdB, PvD and MAvE planned the review and wrote the initial draft. TW, MRB, MS, TP, MS, PM, MvdB, GMH, LHvdB, MS, PvD and MK provided critical review, edited the manuscript and approved the final version. SV created figures 1 and 2. TP provided figure 3.
Funding Funding support from the National Health and Medical Research Council of Australia (Project grant numbers 510233, 1024915, 1055778; Program Grant #1132524, Dementia Research Team Grant #1095127 and Partnership Project #1153439) and Motor Neuron Disease Research Institute of Australia is gratefully acknowledged. MK was supported by an NHMRC Practitioner Fellowship (#1156093). PvD holds a senior clinical Investigatorship of FWO-Vlaanderen and is supported by the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders, the ALS Liga België and the KU Leuven funds “Een Hart voor ALS”, “Laeversfonds voor ALS Onderzoek” and the “Valéry Perrier Race against ALS Fund”. Several authors of this publication are member of the European Reference Network for Rare Neuromuscular Diseases (ERN-NMD).
Competing interests MAvE received grants from the Netherlands Organization for Health Research and Development (Veni scheme), The Thierry Latran foundation and the Netherlands ALS foundation (Stichting ALS Nederland), the MND association, FIGHT-MND and the EU Joint Programme-Neurodegenerative Disease Research (JPND), has consulted for Biogen and serves on the medical ethical review board of the UMC Utrecht.
Patient consent for publication Not required.
Provenance and peer review Commissioned; externally peer reviewed.