Article Text

Abstract
Background The genetic drivers of multiple sclerosis (MS) clinical course are essentially unknown with limited data arising from severity and clinical phenotype analyses in genome-wide association studies.
Methods Prospective cohort study of 127 first demyelinating events with genotype data, where 116 MS risk-associated single nucleotide polymorphisms (SNPs) were assessed as predictors of conversion to MS, relapse and annualised disability progression (Expanded Disability Status Scale, EDSS) up to 5-year review (ΔEDSS). Survival analysis was used to test for predictors of MS and relapse, and linear regression for disability progression. The top 7 SNPs predicting MS/relapse and disability progression were evaluated as a cumulative genetic risk score (CGRS).
Results We identified 2 non-human leucocyte antigen (HLA; rs12599600 and rs1021156) and 1 HLA (rs9266773) SNP predicting both MS and relapse risk. Additionally, 3 non-HLA SNPs predicted only conversion to MS; 1 HLA and 2 non-HLA SNPs predicted only relapse; and 7 non-HLA SNPs predicted ΔEDSS. The CGRS significantly predicted MS and relapse in a significant, dose-dependent manner: those having ≥5 risk genotypes had a 6-fold greater risk of converting to MS and relapse compared with those with ≤2. The CGRS for ΔEDSS was also significant: those carrying ≥6 risk genotypes progressed at 0.48 EDSS points per year faster compared with those with ≤2, and the CGRS model explained 32% of the variance in disability in this study cohort.
Conclusions These data strongly suggest that MS genetic risk variants significantly influence MS clinical course and that this effect is polygenic.
Statistics from Altmetric.com
Introduction
During the past few decades, using candidate gene, linkage studies and genome-wide association study (GWAS) approaches, at least six human leucocyte antigen (HLA) loci and 110 non-HLA genetic loci have been identified as associated with multiple sclerosis (MS) onset.1–6 In contrast, there has been comparatively less work into the genetic drivers of MS clinical course. The large GWAS have shown no significant loci that differentiate progressive-onset MS from bout-onset MS, even in cohorts enriched for progressive cases.7 Similarly, no association has been found with disability.4 ,5 ,8 ,9 This most likely reflects the comparative difficulty in evaluating clinical course in genetic studies, since MS clinical course (conversion to active disease, relapse or disability progression) is not easily studied by GWAS, as GWAS are cross-sectional or case–control in design, while MS clinical course is best assessed longitudinally, and ideally in real time and from disease onset, so as to reduce potential impacts of reverse causality or heterogeneity by treatment or other disease aspects.
A more expeditious approach to assess genetic determinants of clinical course is to use established GWAS determined MS onset associated variants, and evaluate these as predictors of MS clinical course in a prospective longitudinal cohort study, as we can hypothesise that those genetic variants associated with MS onset are potentially also involved in clinical course. This brings to bear the strengths of this study design, while mitigating the power limitations attendant on using a genome-wide approach.
Previously, using this approach in a well-described longitudinal cohort of established MS cases, we have shown some evidence that known MS risk single nucleotide polymorphisms (SNPs) influence relapse and disability.10 Here, we extend this approach to analyse data from a prospective cohort of cases recruited around their first clinical episode suggestive of central nervous system (CNS) inflammatory demyelination referred to as a first demyelinating event (FDE), and followed for 5 years with repeated neurological review. All measures of MS clinical course have been collected prospectively, including conversion to MS, relapse and measures of disability.
Methods
Study design
The Ausimmune case–control study11 was designed to elucidate environmental and genetic risk factors for the onset and early progression of MS. The Ausimmune study recruited a study sample of 282 case participants with a first clinical diagnosis of CNS demyelination indicating a high risk of developing MS. Case participants in the Ausimmune study have been followed up in the AusLong cohort study (the analyses presented here) including follow-up to 5 years from study recruitment (84.6% retention).
The AusLong study cohort included in these analyses is slightly different from the original Ausimmune study case participant sample, as a result of clinical information provided up to the 5-year review. Three Ausimmune study case participants were identified as not having had a MS-associated FDE (one neuromyelitis optica, one Susac's syndrome and one pineal germinoma). Additionally, three cases originally regarded as bout onset were reclassified as being progressive onset after follow-up.
The Ausimmune and AusLong studies were approved by nine regional Human Research Ethics Committees. All participants gave written informed consent.
Measurement of clinical outcomes
Several clinical outcomes were evaluated, including time to conversion to definite MS, number of relapses and annualised disability progression from FDE to 5-year review (average 5.8 years from onset). Conversion to MS was defined primarily as the occurrence of two or more clinical demyelinating episodes, thus satisfying the diagnostic requirements of dissemination in space and time, or a single episode plus paraclinical evidence, as per the 2005 McDonald criteria12 (a minority of cases were diagnosed following MRI (either at the 2/3-year or 5-year reviews) based on this latter criterion (n=20)). Conversion to MS was reported at annual review and cross-checked with neurological records. A relapse was defined according to the 2001 McDonald criteria13 as the acute or subacute appearance or reappearance of a neurological abnormality (lasting at least 24 hours) in the absence of other potential explanatory factors. Relapses were reported at annual review and only relapses which were diagnosed and verified by a neurologist were included in this analysis. Disability was assessed by the Kurtzke Expanded Disability Status Scale (EDSS)14 assessed at the 5-year review by the study neurologists.
Genotyping and SNP selection
DNA from AusLong participants was genotyped using the Illumina Human Exome BeadChip (Illumina Human Exome-12 v1.2 array), which includes ∼244 000 exome SNPs with an additional ∼87 000 MS relevant variants added as a customised component. Quality control15 was conducted based on previous protocols. In general, individuals were excluded based on the following criteria: a call rate of <99%, gender error or duplicate discordance. Variants were excluded on the basis of a call rate of <99% or a deviation from the Hardy-Weinberg equilibrium with p<1.0×10−6. Principal components analysis was carried out to identify population outliers.16 All samples were identified as Caucasian and no outliers were identified to suggest that population stratification was influencing the results. Data on the previously published 110 MS-associated non-HLA region SNPs1 ,2 ,7 and 6 HLA SNPs2 ,3 ,17 were extracted for analysis. For non-HLA proxies SNP selection, we set the threshold at R2≥0.6. For one SNP rs6498184, we selected the nearest SNP rs12599600 (DPrime=1) as a proxy. The six HLA SNPs assessed were rs3135391 (HLA-DRB1*15:01), rs4713274 (HLA-A*02:01), rs1059615 (DRB1*03:01), rs9277561 (rs9277565_T), rs9266773 (HLA-B*44:02), rs7775055 (HLA-DRB1*08:01).
Data analysis
Predictors of time to conversion to MS and to relapse were evaluated by Cox proportional hazards regression models, the latter for repeated events using the gap-time model by Prentice et al.18 All covariates satisfied the proportional hazards assumption.
While the total study sample was 279 participants, the analyses in this paper are restricted to the 127 cases with a classic FDE and genotyping data for MS/relapse and 125 cases for disability progression.
Annualised change in EDSS (ΔEDSS) was calculated by taking the 5-year review EDSS and dividing by the duration between the day before the date of the FDE (EDSS assumed to be 0) and the 5-year review; this proportion was rendered into an annualised value. Since EDSS was assumed to be 0 on the day before FDE in our models, we did not adjust for baseline EDSS. No case reported prior neurological disability or symptoms. Predictors of ΔEDSS were evaluated using linear regression, adjusted for whether persons were having a relapse at the time of their 5-year EDSS assessment. Since the annualised change in disability was highly skewed, a log transformation was applied to satisfy linear regression assumptions. Residuals for the EDSS outcome were near normally distributed after log transformation and met criteria for minimal heteroscedasticity. All means and coefficients, however, were back-transformed and presented on the original scale of ΔEDSS. As for covariate selection, the core model was adjusted for age, sex and study site, and these covariates were selected for the relevance of age and sex in MS, while study site was an appropriate covariate due to the multicentre nature of the study. Age, sex and study site were identified as a true confounder in our MS/relapse model. Age, sex, study site and whether participants were having a relapse at the time of their 5-year disability measurement were identified as true confounders in the disability analysis. Regarding treatment with disease-modifying therapies, very few cases (<2%) received treatment after FDE, but it was near universally applied after MS, although using treatment status in the model did not significantly alter the findings. Therefore, treatment status was not included as a confounder. Adjustment for Bonferroni multiple comparisons was applied for 116 SNPs (110 non-HLA and 6 HLA), this defined as the as-measured p value multiplied by the number of tests (n=116).19
We created a cumulative genetic risk score (CGRS) which included the significant SNPs from the MS/relapse analysis and the ΔEDSS analysis separately. We created two variables that provided values for the number of risk genotypes affecting outcomes, to represent two CGRS.20–22 For example, those participants with three, four or five genotypes that associated with higher probability of conversion to MS were each compared with the reference group—those carrying fewer than two associated SNPs. Where only the homozygous level of the risk genotype was significantly associated with outcomes, this was defined as the risk genotype, but where both the heterozygote and homozygote carriers of the risk genotypes were significantly associated with outcomes, these were defined as the risk genotypes.
To assess potential type 1 error and provide additional evidence to support that our findings did reflect altered risk of the outcome, we undertook a simulation involving the 3 HLA SNPs and 14 SNPs found to significantly predict MS/relapse and disability progression (7 for MS/relapse, 7 for disability progression; see online supplementary tables S1 and S2). For this analysis, a permutation simulation was done where AusLong participants' genotype data for these SNPs were randomly reallocated in equivalent proportions of genotype to that in the original sample. For example, the proportions of genotype rs842639 were such that 125 persons had the reference genotype and the remainder the non-reference genotype (table 3). The simulated genotypes were generated, analysed and the magnitudes of the estimates resultant therefrom retained. These simulations were run 50 000 times and the proportion of magnitudes resulting that were as or more extreme than that found in the as-measured analyses denoted the significance for each SNP.
supplementary tables
All statistical analyses above were conducted in Stata/SE V.12.1 (StataCorp LP, College Station, Texas, USA).
Results
Characteristics of participants
Of the 279 participants in the AusLong study, genotype data were available for 207 participants; 127 of these had a classic FDE and were evaluated in our analyses. Of these, 98 (77.2%) were female and the mean age at study entry was 37.8 (SD 9.5) years. Sixty-eight (53.5%) had converted to MS by 5-year review and had 151 relapses, while the median EDSS at 5 years was 1 (IQR 0–2).
Non-HLA SNP predictors of clinical outcomes
We identified five non-HLA SNPs which predicted conversion to MS, while four non-HLA SNPs predicted relapse (table 1). Two SNPs (rs1021156 near PKIA and ZC2HC1A, rs12599600 near PRM1 and RMI2) were associated with both MS and relapse. None of the SNPs which predicted conversion to MS and/or relapse showed any association with ΔEDSS. While none of these associations persisted in significance on adjustment for multiple comparisons (116 tests), the consistent effect direction between conversion to MS and relapse, even for those SNPs that did not significantly associate with the other outcome, increases our confidence that the associations are genuine.
Seven top non-HLA-SNPs and their associations with the hazard of conversion to MS and relapse*
Combining the seven SNPs that predicted conversion to MS and/or relapse (table 1) into a CGRS, we found evidence of a significant positive association of increasing number of risk genotypes and subsequent hazard of MS and relapse (table 2, figure 1). While the associations were not neatly dose-dependent for MS or relapse, these results suggest that an increasing number of risk genotypes is deleterious for subsequent disease activity.
Cumulative risk of MS and relapse for the seven SNPs associated with conversion to MS and relapse
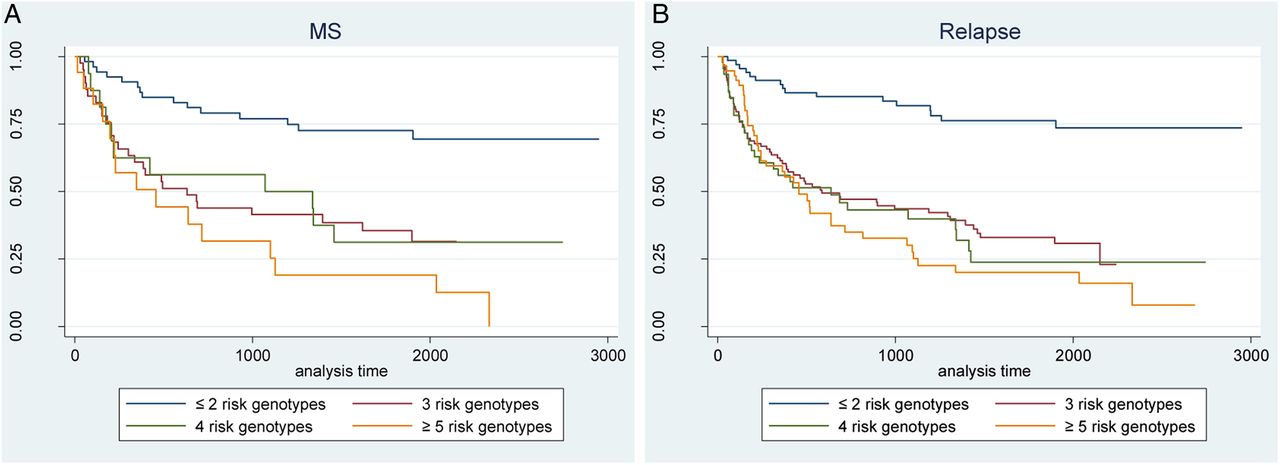
Kaplan-Meier survival plot for cumulative genetic risk score of conversion to MS (A) and relapse (B). MS, multiple sclerosis.
SNP predictors of annualised change in disability
We identified seven non-HLA SNPs (table 3) that were associated with ΔEDSS; no HLA SNPs significantly predicted disability progression. None of these SNPs showed any material association with conversion to MS or relapse.
Seven top SNPs and their associations with annualised change in disability (ΔEDSS)*
For the seven disability-associated SNPs, where the risk genotype was the genotype associated with an increase in EDSS and not necessarily the minor allele, we found a strong and significant dose–response (table 4, figure 2). For example, compared with those with ≤2 risk genotypes, those with ≥6 risk genotypes had an annual disability progression rate of nearly 0.5 EDSS points greater, which over 5 years equates to 2.5 EDSS points. The CGRS model explained 32.7% of the variance in disability progression (R2=0.327, ptrend=1.53×10−9)
Cumulative risk of disability for the seven SNPs that predicted ΔEDSS

{kind=link}
{kind=link}
The line plot of cumulative genetic risk score predicting ΔEDSS. Results presented as geometric mean ΔEDSS and 95% CI. ΔEDSS, annualised disability progression from FDE to 5-year review; EDSS, Expanded Disability Status Scale; FDE, first demyelinating event.
HLA SNP predictors of MS clinical course
In addition to the 110 non-HLA MS-risk associated SNPs, we also examined the association of six HLA SNPs which have been associated with MS risk. These results show that only two of these were associated with the hazard of MS and relapse, but not disability, while the prototypical HLA MS risk-associated loci, DRB1*1501, was not significantly associated with any clinical outcomes in this study (table 5). Following permutation analysis (see online supplementary table S1), the association between rs9266773 tagging B*44:02 and relapse became more significant (p=0.0003), persisting after multiple testing.
The association between three HLA SNPs and MS clinical course†
Discussion
Using a longitudinal cohort of participants with a first neurological presentation of symptoms suggestive of CNS demyelination, we investigated whether known MS susceptibility SNPs were associated with MS clinical course and disability progression in early disease. We found that several known MS risk-associated SNPs significantly influenced MS clinical course, including seven SNPs which predicted the hazard of MS and/or relapse and seven other SNPs which predicted ΔEDSS. While none of these SNPs individually remained significant after adjusting for multiple comparisons, epidemiological supports such as dose dependency and internal consistency between related clinical outcomes supported the validity of taking these SNPs forward to a CGRS assessment. The CGRS analysis showed that, in combination, a greater number of risk genotypes had a highly significant positive association with conversion to MS (HR 5.98 for ≥5 risk genotypes vs ≤2 risk genotypes), relapse (HR 6.07 for ≥5 risk genotypes vs ≤2 risk genotypes) and ΔEDSS where the change in EDSS for those who had ≥6 risk genotypes was 0.48 EDSS points per year greater than reference.
Our CGRS model for disability progression explained 32.7% of the variance in MS disability progression within this data set. These results suggest that these seven common variants in combination significantly contribute to disability progression of MS.
The risk variants detected were completely different between disability and MS/relapse. Hence, we hypothesised that MS/relapse and disability progression may be driven by different genetic pathways, with MS and relapse driven by CNS inflammation, whereas disability progression may be more driven by neurodegeneration. Previous work has suggested that the two processes may be independent,23 although this is controversial. The lack of overlap between genetic variants that may drive conversion and relapse and those associated with disability progression is of great interest and may add support to the argument that these two processes may be independent and require different approaches to treatment.
One interesting observation in our study was that the effects on MS clinical course of the HLA SNPs that have such significant effects on MS risk were varied, with only HLA-B*44:02 (rs9266773) having a significant protective association with relapse and conversion to MS, the latter reaching statistical significance on permutation testing after correction for multiple testing. The MS risk allele of HLA DRB1*15:01 was not clearly associated with MS clinical course in this study, supporting findings from some but not all previous studies.2 ,24 In previous work, the MS risk allele of HLA DRB1*15:01 was not associated either with clinical course MS (primary-progressive multiple sclerosis (PPMS) vs relapsing-remitting multiple sclerosis (RRMS))25 or with the severity of MS26–28 but was associated with an earlier age of onset.25 ,27 On the contrary, other studies have shown a significant association between HLA DRB1*15:01 and the severity of MS29 ,30 potentially modulated by ethnicity.
We have shown some overlap with our previous (independent) study in established MS that further validates this work. In particular, the MS risk SNP near the RGS1 gene associated with the hazard of MS in the current analysis was significantly associated with subsequent relapse risk in our previous study.10
Basing results only on statistical significance in a longitudinal MS study when looking at multiple genetic markers is difficult and requires large sample sizes. The major limitation of our study is the small sample size, particularly when this is further reduced by restriction to only those with genotyping data and those with initial bout-onset disease with onset close to the time of study entry. Therefore, in our study, we have also used several other epidemiological concepts to provide support for our results, including dose dependency of allelic effect, internal consistency between related outcome measures (MS and relapse), and external consistency of directionality with associations found previously, as well as CGRSs. All seven SNPs that were associated with MS and relapse risk had significant allele dose–responses, and all effects were in the same direction for the hazard of MS and relapse and in the same direction as for MS risk in GWAS providing support for their significance. These seven SNPs may be near genes that have significant effects on MS clinical course and warrant further investigation.
A key strength of our study is its long follow-up, beginning at the first presentation of symptoms of disease and continuing for at least 5 years from onset. This allows confidence that the clinical course parameters measured are accurate, particularly for disability progression. Large GWAS analyses, while benefiting from a large sample size allowing for the ability to adjust for multiple comparisons, are methodologically limited by their inability to do more than compare groups, or measure progression using cross-sectional measures, rather than using time-to-event prospective analyses of clinical course that we have used in the present study. In this study, we have used the study strengths of a prospective cohort study design and evaluated the known MS risk-associated SNPs as predictors of clinical course. In this fashion, we retain the methodological strengths of the study design, the accuracy of prospective clinical course monitoring and the reduction of reverse causality, while not having the statistical limitations of trying to evaluate using a genome-wide approach. We have used this approach previously in our cohort of established MS (average disease duration 12 years). However, that study was undertaken in a cohort that experienced little disability progression over a mean follow-up of 2.3 years and was in a largely treated population with a low annual relapse rate. Disability was measured at the 5-year face-to-face review where material stable disability accumulation is likely31 and annualised from the day before FDE onset, when it was assumed to be 0 as no participant reported pre-existing neurological disability. Additionally, the disability outcomes were adjusted for relapse status at 5 years as this was found to be a true confounder. However, it is possible that some of the measured 5 year EDSS values may not be sustained as regression can occur in a small subset of MS cases.31 This study makes use of a cohort followed essentially from symptom onset and who accordingly were not on disease-modifying therapy or yet suffering appreciable impacts of disease. Since patients with relapsing–remitting MS have a highly variable time interval between the first and the second episode of CNS demyelination which clinically or radiologically defines the onset of MS.32 Understanding the genetic determinants of this temporal window of disease clinical course is important as this could allow appropriate counselling, open new avenues for drug development and allow better selection from the available treatment options. Even so, our results should be replicated in other longitudinal cohorts to allow greater confidence in their veracity.
In conclusion, our findings support an association between known MS risk genes and MS clinical course. These data support a role for genetic factors in MS progression and suggest that the genetic drivers of MS progression are polygenic. These results require validation in other cohorts, but with replication these loci may serve as potential targets for further translational research.
Acknowledgments
The members of the AusLong Investigator Group are: Robyn M Lucas (National Centre for Epidemiology and Population Health, Canberra), Keith Dear (Duke Kunshan University, Kunshan, China), Anne-Louise Ponsonby and Terry Dwyer (Murdoch Childrens Research Institute, Melbourne, Australia), Ingrid van der Mei, Leigh Blizzard, Steve Simpson Jr and Bruce V Taylor (Menzies Institute for Medical Research, University of Tasmania, Hobart, Australia), Simon Broadley (School of Medicine, Griffith University, Gold Coast Campus, Australia), Trevor Kilpatrick (Centre for Neurosciences, Department of Anatomy and Neuroscience, University of Melbourne, Melbourne, Australia). David Williams and Jeanette Lechner-Scott (University of Newcastle, Newcastle, Australia), Cameron Shaw and Caron Chapman (Barwon Health, Geelong, Australia), Alan Coulthard (University of Queensland, Brisbane, Australia) and Patricia Valery (QIMR Berghofer Medical Research Institute, Brisbane, Australia).
References
Footnotes
A full list of members of the AusLong/Ausimmune Investigator Group is provided in the Acknowledgments.
Contributors GP did the statistical analysis under supervision by SS, BVT and IvdM. GP and SS did the interpretation, and wrote the manuscript, with input from YZ, FW, IvdM, JCC, RL, A-LP and BVT. IvdM, RL, A-LP and BVT conceived and designed the study. All authors revised and approved the final version of the manuscript. BVT had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Funding The AusLong cohort study was supported by the National Health and Medical Research Council of Australia (Grant reference number: 54922).
Competing interests None declared.
Patient consent Obtained.
Ethics approval Nine regional Human Research Ethics Committees.
Provenance and peer review Not commissioned; externally peer reviewed.