Article Text

Abstract
Objective: To determine the relation of amyloid and tau pathology in the hippocampal formation to decline in memory and other cognitive functions in Alzheimer’s disease (AD).
Methods: Regression models were used to relate semiquantitative measurements of amyloid plaques, neurofibrillary tangles (NFTs) and neuropil threads (NTs) at autopsy with antemortem performance in memory, abstract/visuospatial and language domains in two independent samples (n = 41, n = 66) that had repeated neuropsychological measurements before death.
Results: In both groups, the number of NFTs in the entorhinal cortex, subiculum and CA1 region was inversely associated with memory performance at the last visit before death. However, the number of amyloid plaques and NTs in the entorhinal cortex was also inversely related to poor memory function. Moreover, as the number of plaques or NTs increased in any region of the hippocampal formation, there was a more rapid decline in memory performance over time; a similar decline was associated with increasing numbers of NFTs in the CA1 or subiculum. In contrast, there was no association between amyloid plaques, NFTs or NTs in the frontal or parietal lobe and performance in memory, nor was there an association between plaques, NFTs or NTs in the hippocampal formation and cognitive functions unrelated to memory.
Discussion: This study implicates both amyloid deposition and tau pathology in the hippocampus as an early and late cause of decline in memory function over time in AD. Memory performance appears to be specifically related to the amount of amyloid plaques, NFTs and NTs in the entorhinal cortex and hippocampus.
Statistics from Altmetric.com
Alzheimer’s disease (AD) is characterised by progressive cognitive deterioration leading to impairment in activities of daily living. Neuropathological key features in the brain are amyloid plaques, neurofibrillary tangles (NFTs) and neuropil threads (NTs). Amyloid plaques consist of fibrils formed from the amyloid-β (Aβ) peptide accompanied by microgliosis, dystrophic neuritis and astrocytic processes. NFTs and NTs are intraneuronal protein aggregates arranged in paired helical filaments, formed by hyperphosphorylation of the microtubule associated protein tau.
All three features occur in an increasing number during the progression of AD,1 with prominence in the temporal neocortex, entorhinal cortex and hippocampus in the early phases.1 Degenerative changes in these structures play a major role in the memory dysfunction observed at early stages of AD.2 3 Which underlying abnormality drives the dysfunction remains unknown, and few data are available regarding the relation of NFT formation and amyloid deposition in these structures to specific cognitive functions.
We characterised hippocampal tau and amyloid pathology in the brains of participants of two independent cohorts. Specifically, we quantified NFTs, NTs and amyloid plaques in the entorhinal cortex and hippocampus, explored the degree to which they relate to memory and other cognitive abilities assessed proximate to death, and explored how they relate to decline in cognitive functions over time. We also assessed the association of amyloid plaques, NFTs and NTs in other brain regions with cognitive function to determine the specificity of the observed relationships.
METHODS
Subjects and setting
Participants were part of a longitudinal epidemiological or a clinical cohort. Participants in the epidemiological cohort were from a random sample of Medicare recipients ⩾65 years and residing in northern Manhattan, New York, USA.4 Each participant underwent a standard assessment, including medical history, physical/neurological examination and a neuropsychological battery.5 Participants were recruited at two time periods (1992–1994 and 1999–2002) and followed at 18 month intervals. For this study we used data from 41 participants who died during the study period, had detailed semiquantitative data from brain autopsy and at least two complete assessments prior to death.
The clinical cohort consisted of patients presenting with memory complaints to the Memory Disorders Center at the New York State Psychiatric Institute or Columbia University Medical Center. All patients underwent the same standard assessment described above. Requiring detailed semiquantitative data from brain autopsy and at least two complete assessments prior to death, we included 66 patients in this study. In both cohorts, compared with the excluded participants, those included were older at baseline but there were no differences in sex or ethnicity.
Neuropathological assessment
Neuropathological assessment was performed blinded to the clinical data. One half-brain was assessed grossly while it was dissected at the fresh state to harvest blocks which were deep frozen and banked. The contralateral half brain was immersed in 10% buffered formalin phosphate solution for neuropathological evaluation, as described previously.6 The neuropathological diagnosis of AD was based on CERAD criteria.7 The likelihood that dementia was due to the Alzheimer changes was assessed according to the National Institute on Aging–Reagan Institute criteria.8 A Braak stage was assigned to reflect the extent of involvement of NFT.9 Briefly, a minimum of 18 standardised paraffin embedded blocks were obtained from each fixed half-brain. Sections (7 μm thick) from all blocks were stained with Luxol fast blue and counterstained with haematoxylin–eosin for general survey. Selected sections were stained with Bielschowsky for evaluation of axons, amyloid plaques, and NFT and glial tangles; antibodies against β-amyloid for vascular and parenchymal deposits (Dako M0872; Dako North America Inc, Carpinteria, California, USA); phosphorylated tau (AT8) for neuronal/glial tangles and neuropil threads; ubiquitin for ubiquitinated cytoplasmic, nuclear or axonal aggregates; and α-synuclein for Lewy bodies, Lewy neurites and glial tangles.
Plaques (neuritic, immature and diffuse), NFT and NTs were assessed semiquantitatively within the following regions: (a) anterior frontal (including Brodmann area (BA) 9); (b) posterior frontal/paracentral (BA4 and BA1, 3, 2, 5, 40, 7); (c) occipital (BA18, 17, 32); (d) hippocampal formation at the level of the lateral geniculate body (including BA28, 36, 20); (e) amygdaloid nucleus (including rostral BA28); (f) temporal pole and pre-pole (BA38, 20, 21, 22); and (g) head hippocampal formation (including BA28, 36, 20).
The mean number of amyloid plaques in five random 100× fields per slide was recorded using Bielschowsky stained sections or the sections subjected to β-amyloid antibodies. Amyloid burden was graded on a 4 point scale (0 = no deposit; 1+ = 1–5 plaques; 2+ = 6–15 plaques; 3+ = >15 plaques). The mean number of NFTs in five random 100× fields per slide was recorded using sections subjected to AT8 antibodies, and ranked as follows: none = no NFTs; mild = 1–6 NFTs; moderate = 7–15 NFTs; severe = >15 NFTs. Sections subjected to AT8 antibodies were used to evaluate the density of NTs which was likewise graded on a scale from “none” to “severe”. Figure 1 shows a representative of tau and amyloid pathology in the hippocampus.

Tau and amyloid pathology in the hippocampus. Bielschowsky, original magnification 100×.
Neuropsychological assessment
Orientation was evaluated using the Mini-Mental State Examination.10 Language was assessed using the Boston Naming Test,11 Controlled Word Association Test,12 category naming and the Complex Ideational Material, and the Boston Diagnostic Aphasia Evaluation.13 Abstract reasoning was evaluated using the Wechsler Adult Intelligence Scale-Revised Similarities subtest14 and the non-verbal Identities and Oddities subtest of the Mattis Dementia Rating Scale.15 Visuospatial ability was examined using the Rosen Drawing Test16 and the Benton Visual Retention Test.17 Memory was evaluated using the Benton Visual Retention Test17 and the Selective Reminding Test18 (total recall, long term recall, long term storage, continuous long term storage, words recalled on last trial, delayed recall, delayed recognition). A consensus committee comprising neurologists, neuropsychologists and psychiatrists reviewed the results from all examinations and assigned individuals to one of three categories: normal cognitive function, cognitive impairment without dementia or dementia. Dementia was defined by DSM-IV criteria19 and required cognitive impairment in several domains and functional impairment (Clinical Dementia Rating ⩾1).11 Cognitive impairment without dementia was diagnosed in participants who had abnormal results in cognitive tests but had no significant cognitive impairment (Clinical Dementia Rating 0.5).
APOE genotype
Apolipoprotein E (APOE) genotypes were determined as described by Hixson and Vernier with slight modification,20 and classified as having no APOEϵ4 allele (−/−) or at least one APOEϵ4 allele (−/4 or 4/4).
Statistical methods
We transformed each neuropsychological test into z scores for the purposes of standardisation and conducted a factor analysis using a principal component analysis with varimax rotation and Kaiser normalisation. This resulted in three factors of which the contributing tests were mutually exclusive: (1) a memory factor, in which the seven subtests of the Selective Reminding Test were the main contributors; (2) a visuospatial/cognitive factor, where visuospatial and tests of reasoning were the main contributors; and (3) a language factor, in which language measures from the Boston Naming Test,11 Controlled Oral Word Association Test12 and the Wechsler Adult Intelligence Scale-Revised Similarities14 were the main contributors. We calculated cognitive scores for each participant at each visit by adding the scores of the measures that contributed most to each factor (tests with correlations ⩾0.5).
We used linear regression to relate semiquantitative measures of amyloid plaques, NFTs and NTs in the entorhinal cortex and hippocampal subregions to performance in memory, abstract/visuospatial and language domains at the last visit before death. We adjusted all models for age and sex, then additionally for APOE genotype and education.
We analysed the change in factor scores over time before death by applying generalised estimating equations with repeated measures. The dependent variables were the calculated cognitive scores and the independent variables were the semiquantitative measures of amyloid plaques, NFTs and NTs in the entorhinal cortex and individual hippocampal subregions, time (duration of follow-up) and interaction of amyloid plaques, NFTs or NTs with time. Neuropathological changes were examined as dichotomised variables (present/absent). Sex, age, education and APOE genotype were included as covariates.
The generalised estimating equations analysis yields three main coefficients of interest. A significant p value for the coefficient comparing the groups with/without plaques, NFTs or NTs at baseline indicates a difference between these two groups at baseline. A significant p value for the coefficient of time indicates a significant change in a cognitive score over the total duration of antemortem follow-up. A significant p value for the interaction coefficient of time×plaque/tau group indicates a difference in the rate of change in a factor score depending on the presence/absence of plaques, NFTs or NTs at autopsy; this is the main variable of interest in these analyses.
To explore whether the effects of the individual regions on memory performance were independent, we repeated all analyses adding separately variables for plaque, NFT and NT counts to the regression models that contained all possible clusters of hippocampal regions. For amyloid plaques this variable was coded: 1 = isolated plaques in Ca1, 2 = isolated plaques in subiculum, 3 = isolated plaques in entorhinal cortex, 4 = plaques in Ca1+subiculum, 5 = plaques in Ca1+entorhinal cortex, 6 = plaques in subiculum+entorhinal cortex, 7 = plaques in Ca1+subiculum+entorhinal cortex and 8 = no plaques in any region (reference group). For NFTs and NTs the variable was constructed similarly.
To assess whether observed relations between neuropathological changes and cognitive performance are specific to the hippocampal formation, we repeated all analyses relating plaques, NFT and NT in the frontal/parietal lobes with cognitive performance. We also compared the relation between plaques/NFTs/NTs and cognitive performance between Braak stages I–IV, in which the hippocampal formation is affected, with Braak stages V/VI, in which the disease has further spread to the neocortex.1
RESULTS
The characteristics of the study samples are shown in table 1.
Relation of amyloid/tau pathology to cognitive function at last visit before death
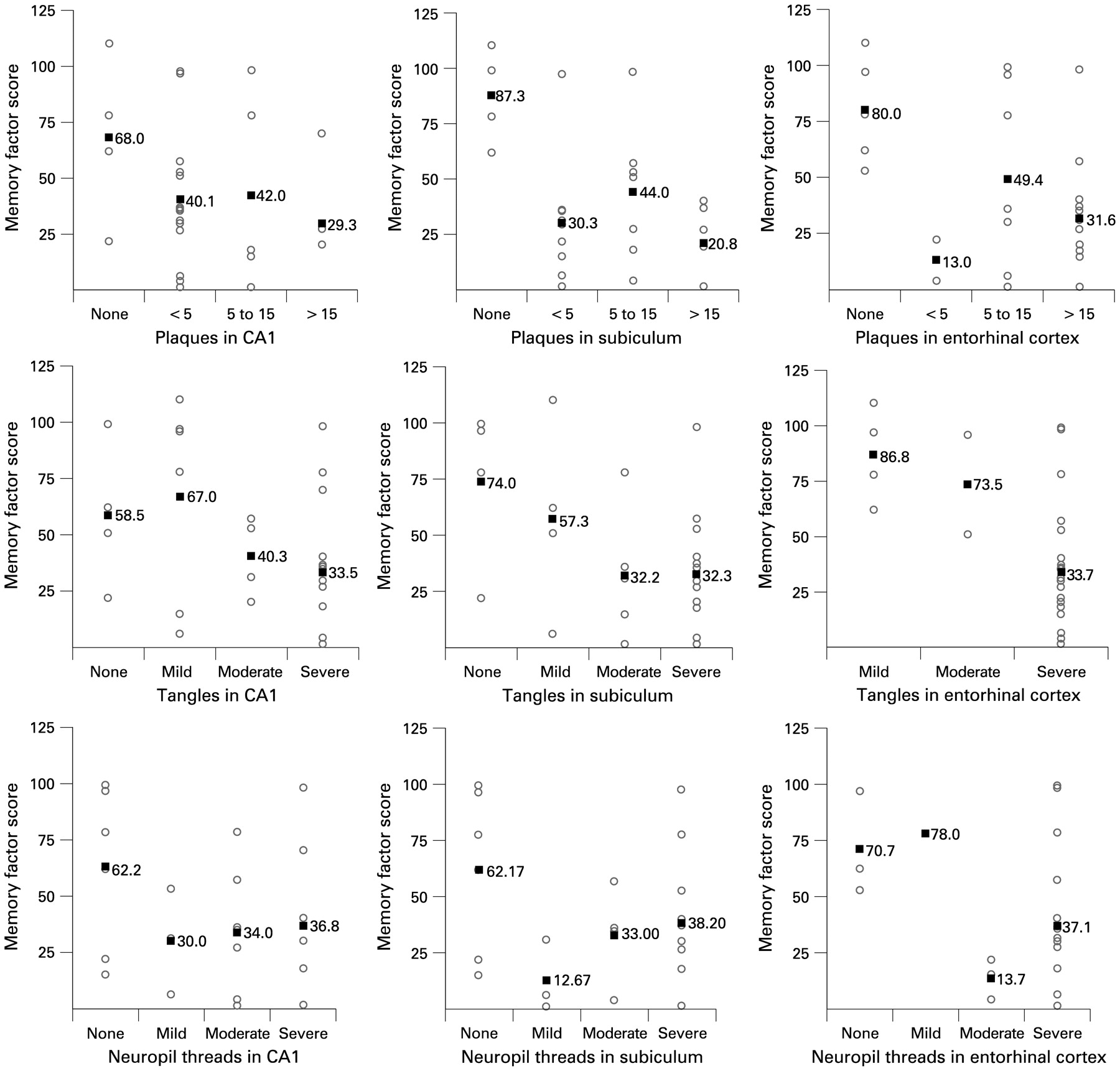
In both samples, the number of NFTs in the entorhinal cortex (p = 0.04, p = 0.03), CA1 (p = 0.01, p = 0.04) and subiculum (p = 0.02, p = 0.04) was inversely associated with memory performance at the last visit before death (figs 2, 3). The number of amyloid plaques (p = 0.03, p = 0.05) and NTs (p = 0.04, p = 0.05) in the entorhinal cortex was also associated with worse memory function.
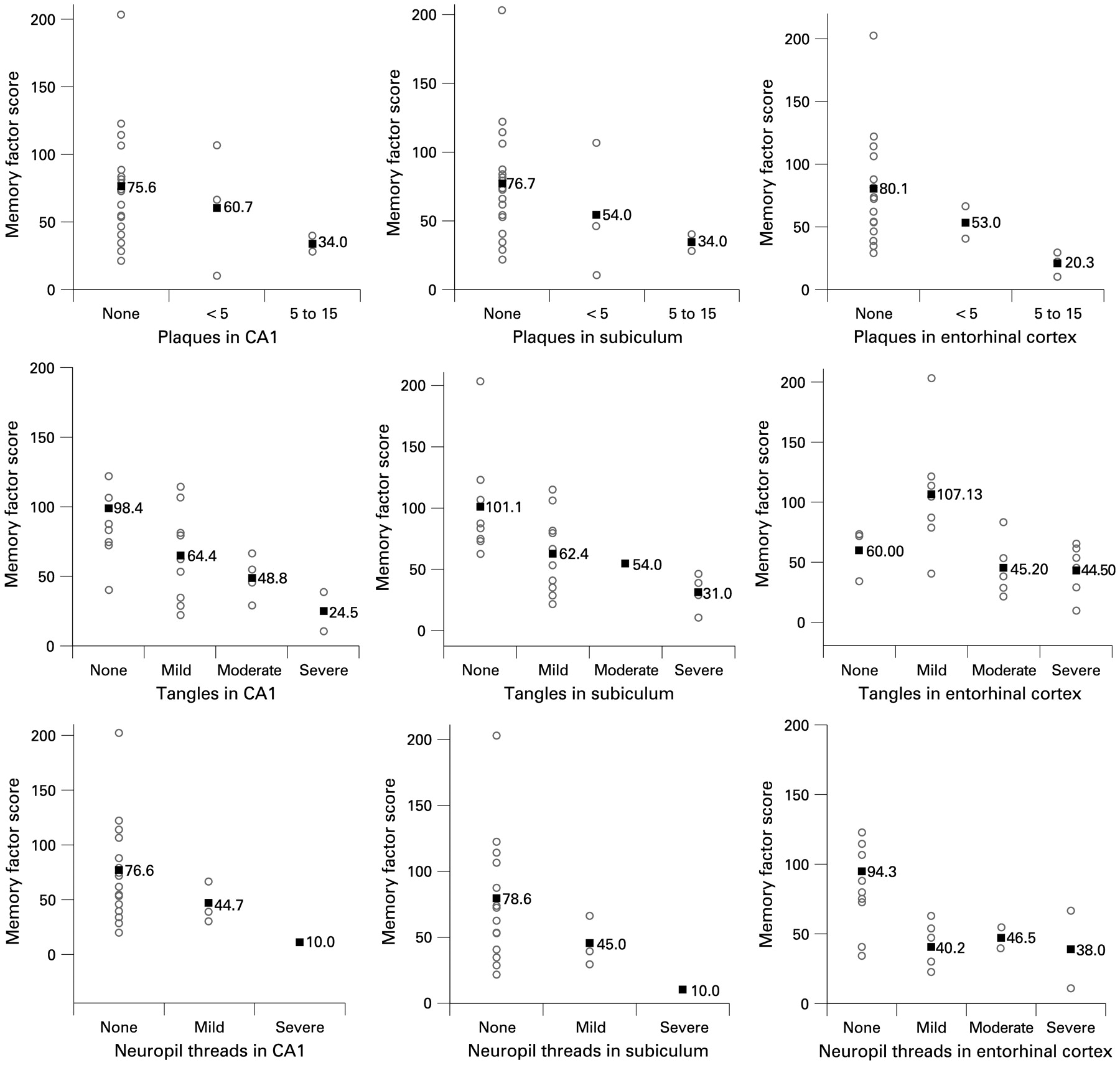
Scatterplots showing the relation of amyloid plaques, neurofibrillary tangles and neuropil threads with memory performance at the last antemortem visit in the epidemiological sample. Numbers in the graph show the means of memory performance scores.
{kind=link}
{kind=link}
{kind=link}
Scatterplots showing the relation of amyloid plaques, neurofibrillary tangles and neuropil threads with memory performance at the last antemortem visit in the clinical sample. Numbers in the graph show the means of memory performance scores.
In the clinical sample, NFTs in the subiculum (p = 0.04) and entorhinal cortex (p = 0.05) were also associated with lower performance in abstract/visuospatial and language domains (data available on request). However, there was no relation in the epidemiological sample, and there was no association between (para)hippocampal amyloid or NT pathology and impairment in non-memory domains in either cohort.
In the analyses relating Braak stage or counts of plaques, NFT and NT in the frontal and parietal lobes with cognitive performance, none of the neuropathological measures was associated with memory or any other cognitive domain. When we repeated all analyses excluding APOE as a covariate, or stratifying by staining technique for amyloid plaques (Bielschowsky staining or Aβ-antibodies), the significance of all results remained unchanged.
Change in memory performance over time
Memory, abstract/visuospatial and language performance declined in both samples over the antemortem follow-up period. In the epidemiological sample, occurrence of plaques or NTs in any region of the hippocampal formation and occurrence of NFTs in the CA1 or subiculum was associated with a significantly faster decline in memory performance over time than absence of these changes (table 2). In the clinical sample, there was no association between neuropathological measures and change in memory function over the follow-up period (data not shown). However, in the clinical sample, the presence of plaques in any assessed region (Ca1: β = −41.4, SE = 17.7, p = 0.01; subiculum: β = −45.75, SE = 18.93, p = 0.01; entorhinal cortex: β = −53.32, SE = 19.75, p<0.007), NTs in any region (Ca1: β = −57.6, SE = 16.6, p = 0.001; subiculum: β = −55.0, SE = 15.8, p<0.0001; entorhinal cortex: β = −55.9, SE = 26.6, p<0.03) and presence of NFT in Ca1 (β = −67.4, SE = 17.7, p = 0.007) was associated with lower memory performance at baseline. Neither plaques, NFTs nor NT in the frontal or parietal lobes were associated with changes in memory or other cognitive domains over time. The significance of all results remained unchanged when age instead of duration of follow-up was used as the time variable, when APOE genotype was excluded as a covariate or when the analyses were stratified by staining technique for amyloid plaques. All models were corrected for multiple testing.
Independence of effects across (para)hippocampal regions
When we added separately the cluster variables for plaques, NFTs and NTs in subregions of the hippocampal formation to the regression models, the effects of amyloid plaques on memory function were additive across all regions in both cohorts. The strongest individual effect was in the entorhinal cortex (β = −52.34, SE = 18.42, p = 0.04). For NTs the effect appeared also additive and was strongest when NTs were present in all regions (β = −56.00, SE = 16.25, p = 0.06). For NFTs, the effect appeared strongest for the subiculum.
DISCUSSION
The accumulation of amyloid plaques in any region of the hippocampal formation was directly related to the decline in memory performance over time. In addition, an increasing number of plaques in the entorhinal cortex was associated with worse memory performance at the last visit before death. NFTs and NTs in the entorhinal cortex, CA1 or subiculum, and the number of NTs in the entorhinal cortex were also inversely associated with memory function and with a faster decline in memory performance over time. There was no association between Braak stage or amyloid plaques, NFTs or NTs in other regions of the brain (such as the frontal or parietal lobe) and memory function. Conversely, there was no consistent relation between any neuropathological measure in the hippocampal formation and non-memory abilities.
The importance of the hippocampal formation for memory function has been established in selective lesion experiments in animal studies.21 22 The hippocampus is central to the formation of new memories and memory consolidation.23 The entorhinal cortex relays multimodal processed information from the sensory cortical areas to the hippocampus, and information processed by the hippocampus to permanent storage sites in the neocortex.24 Dysfunction of these regions leads to impairment in several types of memory, including spatial and recognition memory and operant learning.21 22
Based on these data and the vulnerability of these cortices for the accumulation of tau and amyloid pathology,25 it has been presumed that changes in tau and amyloid contribute to memory deficit in AD. Numerous studies have demonstrated relationships between diagnosis of dementia and global indices of cognitive function and tau26 27 or amyloid pathology.26 28–30 In a study by Garcia-Sierra and colleagues,31 immunoreactivity of phosphorylated tau (AT8) in the perforant pathway correlated with performance on the Global Deterioration Scale. Few studies have explored a more specific relationship between neuropathological findings and memory impairment, the hallmark neuropsychological disturbance of AD. Most of these studies, however, explored either the impact of NFTs but not amyloid pathology on cognition,27 had small sample sizes32 or only global memory assessments.26 28–30 33 34 In addition, most studies assessed memory function solely proximate to death/autopsy26–30 32 33 and did not explore whether observed relations were specific for the assessed brain regions. The present study greatly expands observations from previous studies by illustrating the association between neuropathological changes and longitudinal within subject alterations in cognitive performance. We also showed the specificity of the relationship for the hippocampal formation by comparison with reference regions.
Consistent with previous reports,27 32 NFTs and NTs were associated with memory loss in cross sectional and longitudinal analyses. This supports the hypothesis that tau pathology plays a role in early and late memory decline. In transgenic mice overexpressing tau, tau pathology has been correlated with neuronal dysfunction, destruction of synapses, cortical atrophy and neuronal death.35–38
In contrast with previous studies,28 32 we have shown that amyloid plaques in either region of the hippocampal formation, particularly in the entorhinal cortex, were associated with memory function in cross sectional and longitudinal analyses. This is consistent with the viewpoint that both early and late memory loss in AD is related to Aβ neurotoxicity. It further suggests that within the hippocampal formation, the entorhinal cortex is the subregion predominantly affected by Aβ. Hippocampal functional maps between patients with AD and controls, and J20 transgenic mice and wild-type mice,39 support this observation. J20 mice develop Aβ related neurotoxicity by expressing mutations in the amyloid precursor protein, without developing neuronal cell death or tau pathology. They subsequently develop hippocampal dysfunction. In the respective study, among all hippocampal subregions, the entorhinal cortex was the dominant site of dysfunction observed in both humans and J20 mice. Flurbiprofen, a drug that ameliorates Aβ related neurotoxicity, in turn rescued entorhinal cortex dysfunction.39 Injection of Aβ into the entorhinal cortex of rats results in selective impairments in memory.40
An advantage of our study over previous investigations is its longitudinal design and the use of specific cognitive measures. We used a neuropsychological test battery especially designed for detection of cognitive impairment. Another explanation for the discrepancies between our and previous studies concerning the role of amyloid plaques in memory function is the differences in the interval between the last neuropsychological assessment and death. If the interval is relatively large, the neuropsychological measure may not correctly reflect the impact of pathological changes detected at autopsy. In the present study, the mean length (SD) of the interval between last neuropsychological assessment and autopsy was 2.4 (1.7) years in the clinical cohort and 2.3 (1.6) years in the epidemiological cohort. The fact that we observed consistent relations between amyloid plaques and memory performance in two independent samples and in cross sectional and longitudinal analyses is reassuring. The lack of an association between neuropathological measures and memory performance over time in our clinical sample compared with the epidemiological sample is explained by the fact that the epidemiological sample was recruited from the general community whereas the clinical sample consisted of patients presenting to a clinic because of failing memory. Patients in the clinical sample had significantly lower memory scores than participants in the epidemiological cohort at baseline. Furthermore, in the clinical sample, presence of plaques or NTs in the hippocampal formation and presence of NFTs in CA1 was associated with significantly lower performance in memory at baseline suggesting that amyloid plaques, NTFs and NTs had already led to a severe decline in this domain.
It has been suggested that neither plaques nor tangles but their precedents—soluble tau and Aβ42 oligomers—are responsible for the loss of neurons and memory impairment, and that NFT and amyloid plaques in fact represent a protective neuronal response aimed at sequestering mutant tau and Aβ42. Although we cannot exclude this possibility, the fact that plaques, NFTs and NTs were also inversely associated with longitudinal changes in memory makes this an unlikely explanation.
The correlation between hippocampal pathology and cognition was specific for memory function and restricted to the hippocampal formation. We did not observe a consistent correlation between hippocampal pathology and non-memory abilities, and we demonstrated the specificity of the association between pathological measures and memory function for the hippocampal formation by showing that neither Braak stages nor plaques, NFTs or NTs in the frontal/parietal lobes were associated with memory. Within the hippocampal formation, we showed in both samples the strongest individual effect of amyloid pathology for the entorhinal cortex. This selective correlation with memory is consistent with a special role of the entorhinal cortex in memory function.
Mapping quantitative, spatial and temporal patterns of cognitive dysfunction associated with amyloid and tau pathology are important steps towards a better mechanistic understanding of AD. Our data are consistent with animal studies and greatly expand previous human studies showing that amyloid and tau pathology in the hippocampal formation and memory function are in both early and late stages of cognitive decline continuous correlated variables. Clarifying the molecular mechanisms through which tau and amyloid pathology affect hippocampal function and thereby memory is the next critical step.
Acknowledgments
Statistical analysis was carried out by CR and M-XT.
REFERENCES
Footnotes
Competing interests: None.
Funding: This study was supported by grants PO1 AG07232, P50 AG08702 and RR00645 from the National Institute on Aging (Bethesda, Maryland), the Charles S Robertson Memorial Gift for Research in Alzheimer’s disease and the Blanchette Hooker Rockefeller Foundation (New York, New York).
Ethics approval: The study was approved by Columbia University Institutional Review Board.
See Editorial Commentary, p 703
Linked Articles
- Editorial commentary