Article Text

Abstract
Mutations in the dysferlin gene lead to limb girdle muscular dystrophy 2B, Miyoshi myopathy and distal anterior compartment myopathy. A cohort of 36 patients affected by dysferlinopathy is described, in the first UK study of clinical, genetic, pathological and biochemical data. The diagnosis was established by reduction of dysferlin in the muscle biopsy and subsequent mutational analysis of the dysferlin gene. Seventeen mutations were novel; the majority of mutations were small deletions/insertions, and no mutational hotspots were identified. Sixty-one per cent of patients (22 patients) initially presented with limb girdle muscular dystrophy 2B, 31% (11 patients) with a Miyoshi phenotype, one patient with proximodistal mode of onset, one patient with muscle stiffness after exercise and one patient as a symptomatic carrier. A wider range of age of onset was noted than previously reported, with 25% of patients having first symptoms before the age of 13 years. Independent of the initial mode of presentation, in our cohort of patients the gastrocnemius muscle was the most severely affected muscle leading to an inability to stand on tiptoes, and lower limbs were affected more severely than upper limbs. As previous anecdotal evidence on patients affected by dysferlinopathy suggests good muscle prowess before onset of symptoms, we also investigated pre-symptomatic fitness levels of the patients. Fifty-three per cent of the patients were very active and sporty before the onset of symptoms which makes the clinical course of dysferlinopathy unusual within the different forms of muscular dystrophy and provides a challenge to understanding the underlying pathomechanisms in this disease.
- muscle disease
- muscular dystrophy
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
Introduction
Ten years ago the dysferlin gene was cloned and identified as the gene mutated in limb girdle muscular dystrophy (LGMD) 2B, and Miyoshi myopathy (MM).1 2 While LGMD2B is characterised by proximal muscle involvement, patients affected by MM present with distal muscle weakness at onset.1 2 Subsequently, distal anterior compartment myopathy and a proximodistal mode of onset have been described as further phenotypes/modes of presentation in dysferlin deficiency,3–5 and some patients have been reported with hyperCKaemia.5 During the course of the disease, the phenotypes show a considerable amount of overlap so that weakness may extend from proximal to distal muscle groups and vice versa.4 6 The factors responsible for these distinct patterns of presentation are unknown. Therefore, further characterisation of patients with dysferlin deficiency may help to identify possible distinct features within this entity and might provide clues to underlying pathogenetic mechanisms.7
Dysferlin deficient muscular dystrophy is inherited as an autosomal recessive trait, age of onset has been found to be usually young adulthood and the progression to be slow.4 6 8 Overt cardiac pathology appears not to be a common finding in most patients.4 6 However, cardiac impairment in dysferlin deficiency under stress has been shown in the knockout mouse,9 and two studies have demonstrated dilated cardiomyopathy in a total of six patients with dysferlinopathy.10 11 Dysferlin is a 230 kDa transmembrane protein and has been shown to be involved in the process of membrane repair,12 myoblast differentiation,13 T tubulogenesis14 15 and muscle regeneration,16 most likely through mechanisms of membrane organisation. So far this has not been proven but the hypothesis is based on the following findings: the biochemical structure of dysferlin with six C2 domains which are calcium sensitive domains involved in fusion and trafficking of membranes, the homology to fer-1 in C elegans and otoferlin, both proteins important for vesicle fusion, an upregulation of proteins involved in vesicle trafficking,17 a subsarcolemmal accumulation of vesicles below the surface of dysferlin deficient myofibres18 and altered T tubule morphology and orientation on the ultrastructural level.15 Recent evidence also suggests a role for dysferlin in monocyte phagocytosis.19
A unique finding within the spectrum of muscular dystrophies is that the majority of patients with dysferlin deficiency appear to have good muscle strength before onset of symptoms leading to good performance at sports or to the ability to cope well with physically demanding jobs although this is suggested by mainly anecdotal evidence.19 20 This observation contradicts the clinical course of other muscular dystrophies (MDs) where the majority of patients describe some sort of impaired muscle function before onset of overt weakness. This aspect is difficult to address, has not yet been formally assessed but may be a distinct feature in dysferlin deficiency. In this first study on the clinical spectrum of dysferlinopathy in the UK, we present clinical, genetic, pathology and biochemical data on patients with dysferlinopathy, and specifically address the aspect of pre-symptomatic fitness levels.
Materials and methods
Thirty-six patients with dysferlin deficiency from 32 families were assessed retrospectively. The diagnosis of dysferlinopathy was made based on reduction or absence of dysferlin in the muscle biopsy and mutation analysis. Patients were referred to the Newcastle Muscle Centre as part of the National Commissioning Group designated specialised diagnostic service for LGMDs in the UK for examination and further evaluation. During the clinic visits a complete history and family history were taken, and patients were specifically asked about their muscle strength and athletic performance before onset of symptoms, such as their performance at school sports. A detailed muscle assessment was undertaken by the same examiner in 21 patients, and muscle strength scored.21
Immunoanalysis of muscle biopsies
Muscle biopsies were analysed as part of the National Commissioning Group designated specialised diagnostic service for LGMD. Optimised immunohistochemical and multiplex western blot protocols were used for the demonstration and resolution of muscular dystrophy associated proteins, as previously described.22 23 Primary antibodies used for immunohistochemistry were directed against: β-spectrin (RBC2/3D5), β-dystroglycan (43DAG/8D5), C terminus dystrophin (Dy8/6C5) and N terminus dystrophin (Dy10/12B2), α-sarcoglycan (Ad1/20A6), β-sarcoglycan (1/5B1), γ-sarcoglycan (35DAG/21B5), δ-sarcoglycan (3/12C1) (all from Novocastra, Newcastle, UK) and caveolin-3 (Transduction Laboratories, Oxford, UK). Antibodies used for western blotting were: anti-dysferlin (NCL-hamlet exon 53), anti-dystrophin (Dy8/6C5 C-terminus and Dy4/6D3 rod domain), anti-α-sarcoglycan, anti-β-dystroglycan, anti-γ-sarcoglycan, anti-calpain 3 (NCL-CALP-2C4 and NCL-CALP-12A2) and anti-caveolin 3 (Transduction Laboratories). Myosin heavy chain staining on the post-blotted gel was used as loading control.
Evaluation of biopsies
Two independent raters, experienced in reading of muscle biopsies and western blots, evaluated the biopsies (LK, AA). Protein expression on sections and blots was scored as normal +++, slight reduction ++, reduction +, severe reduction ± and absence −. β Spectrin was used as a marker of membrane integrity on immunohistochemistry.
Sequencing
Sequencing analysis was performed using bidirectional fluorescent sequencing of all 55 exons of the dysferlin gene either in Newcastle upon Tyne (Institute of Human Genetics) or in Würzburg (central sequencing facility of the MD-NET, Institute of Human Genetics, University of Würzburg, Germany).
Twenty-two patients with a molecular diagnosis of LGMD2A (Newcastle Muscle Centre) were used as a control group for the assessment of pre-symptomatic fitness levels in dysferlin deficient patients. In these patients, molecular diagnosis had been established via bidirectional fluorescent sequencing of all 24 exons of the calpain 3 gene in Newcastle upon Tyne, UK.
Muscle assessment
Assessment of muscle strength was conducted by one experienced evaluator (ME). Muscle groups that were examined were those that produced neck flexion and extension, shoulder abduction, flexion, horizontal adduction, elbow flexion including brachioradialis and biceps, elbow extension, wrist flexion and extension, finger flexion and extension. In the lower limb, hip flexion, extension, abduction and adduction, knee flexion and extension and in the ankle plantarflexion, dorsiflexion, inversion and eversion. Each muscle group was assessed in several different positions to determine antigravity activity as well as the ability to resist a manually applied force. Strength was graded using an 11 point scale from 5 (normal strength) to 0 (no activity). The same order of assessment was followed in each case. Each patient was also asked to demonstrate their ability to stand on tiptoes as manual resistance cannot sufficiently demonstrate normal strength plantarflexion.
Upper and lower limb score
Muscles of the upper or lower limb were scored on an 11 point numerical scale (0–10) according to the strength grade (also 11 point). The score of the patient was calculated as the per cent of the maximum score achievable.
Statistical analysis
Fisher's exact test for count and the Student's t test were applied. Statistical significance was established when p<0.05.
Results
Muscle immunoanalysis findings
Immunohistochemistry
Dysferlin was either severely reduced or absent on tissue sections with no apparent correlation to the clinical presentation or underlying mutation. β-dystroglycan was reduced in 17% of samples, and caveolin-3 showed reduced sarcolemmal labelling in 25% of cases. No correlation was found between the residual labelling and the phenotype of the patient.
Western blot
Dysferlin bands were reduced or absent in all patients. Neither the type of mutation nor the phenotype at presentation showed a correlation to the amount of dysferlin detectable on blots. Similar to previously reported data,24 25 we found a secondary reduction of calpain 3 in the majority of patients (77% exon 1, 82% exon 8) and a more variable reduction of caveolin-3 (67% in 12 blots probed with anti-Cav-3).
Correlation of phenotype (MM vs LGMD) with inflammation in the muscle biopsy
There was no correlation between the phenotype and the level of inflammation in the muscle biopsy identified by haematoxylin–eosin staining.
Mutation analysis
Molecular diagnosis was established in all patients with 89% (32 patients) harbouring two mutations in the dysferlin gene: 14 patients were homozygous (including one patient with an additional heterozygous mutation) and 18 patients were compound heterozygous. One of the latter patients had three mutations (patient No 13). In 11% (four patients), only one mutation was detected, including one patient who is likely to be a symptomatic carrier. The patients with one mutation only were included on the basis of dysferlin depletion in the muscle biopsy. A summary of the mutations found in our patient cohort, the percentage of different mutations and their location within the dysferlin gene are depicted in table 1 and figure 1. As previously reported, no mutational hot spots were evident, and mutations were spread widely throughout the gene.
Description of presenting phenotype (LGMD2B/MM/PD/muscle stiffness) functional state (walking, aided walking or wheelchair use with the age of the patients at last examination in years and years since onset of symptoms in parentheses), genetic information, and information on physical activities and sporting before onset of symptoms

Molecular analysis of patients studied. (A) Relative frequency of different types of DYSF gene mutations. (B) Localisation of different mutation within the DYSF gene. Numbers represent patients according to table 2. TM, transmembrane domain.
Creatine kinase analysis
Creatine kinase (CK) levels were increased in all patients. Median CK levels were 7000 iU/l (range 300–19000, normal <150 iU/l) at a median disease duration of 8 years (range 0.5–42 years).
Patient data
Clinical data were available from 36 patients from 32 families (18 men and 18 women); data on current functional status were available in 30 patients. One patient with one mutation detected only was believed to present as a symptomatic carrier (patient No 12b) as he is the brother of patient No 12a in whom two compound heterozygous mutations were found. The ethnic origin of the patients was somewhat variable: 78% (28 patients) Caucasian (northern European descent), 17% of patients (six patients) from India/Pakistan and 5% (two patients) were of Arabic descent.
The phenotype at presentation was identified from the patients' history. Sixty-one per cent of patients (22 patients) initially presented with LGMD2B (initial symptoms were proximal lower limb weakness), 11 patients with a Miyoshi phenotype (difficulties standing on tip toes), one patient presented with proximodistal mode of onset and one patient was unique in presenting with muscle stiffness after exercise.
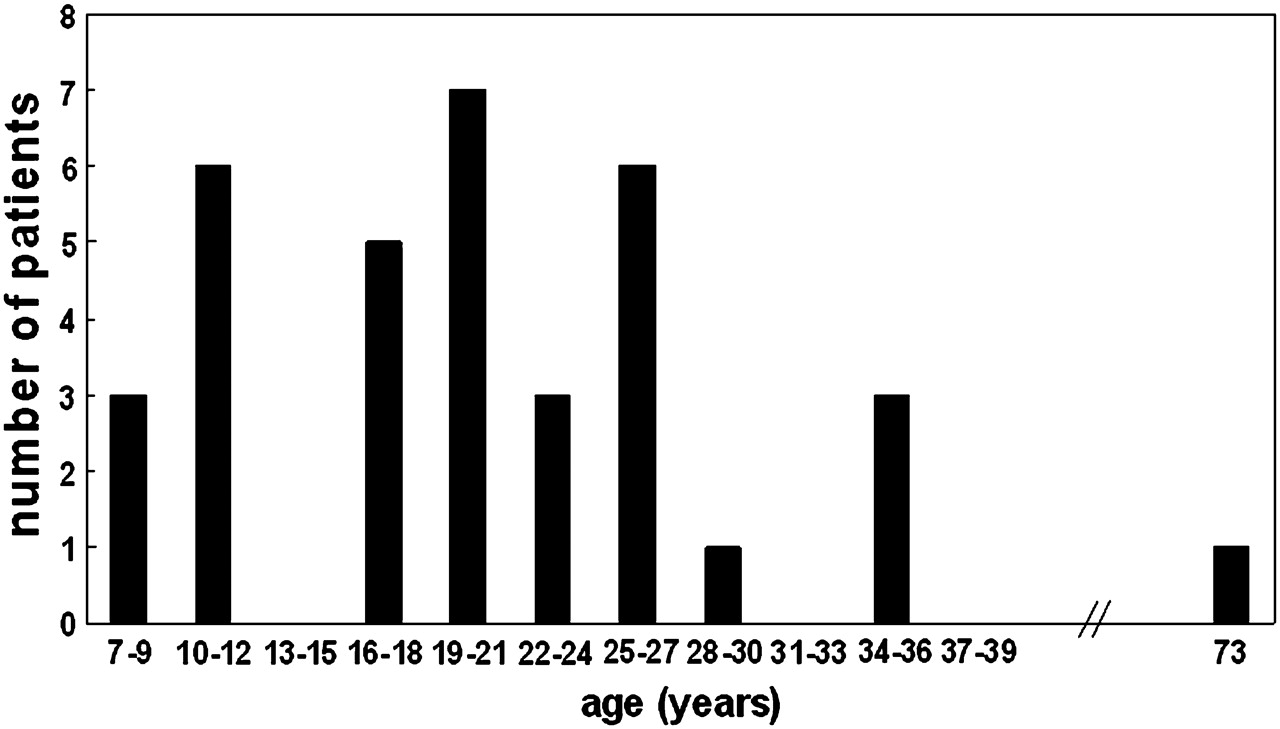
Median age of onset was 20 years (range 7–73 years) and the majority (44%) of patients presented in early adulthood (between 19 and 27 years of age) (figure 2). However, 25% of patients (nine patients) became symptomatic before the age of 13 years. In this group of young patients there was an equal distribution of those patients who were sporty before onset and those who were not (four not sporty and five sporty). At the other end of the spectrum, one patient presented in her eighth decade with muscle stiffness rather than weakness.26 This patient had always been a keen dancer.

Distribution of age of onset in the patients studied. Twenty-five per cent of patients had symptoms before the age of 13 years.
Symptoms at onset
Proximal lower limb weakness in the LGMD2B phenotype and difficulty in toe walking in MM were present in 100% of patients in those groups. Most patients were characterised by a slowly progressive onset of weakness (86% in LGMD2B and 64% in MM). Three women became symptomatic shortly after pregnancy (two with LGMD2B and one with MM), presenting with a more acute onset of weakness. This is a novel finding as we are not aware of any reported patients in whom a possible ‘trigger’ might have influenced the initial presentation of the disease (table 2).
Description of symptoms at initial presentation of the disease
Clinical onset of the disease was similar within the same family with respect to age of onset and presenting phenotype. We did not observe different phenotypes within one family. However, the number of families studied compared with sporadic cases was small (four familial cases).
Muscle function
A detailed assessment of skeletal muscle function was available in 21 patients (figure 3). Median length of disease duration at assessment was 9 years (range 2.5–42 years). The findings reflect disease progression with an overlap of proximal and distal weakness at the time of assessment corroborating that although the diseases may initially present as distinct phenotypes, they largely overlap in later stages. Plantar flexion, hip adduction and hip extension were the weakest muscle groups tested. Hand muscles were relatively well preserved, and scapular winging and contractures were not common features. Asymmetric muscle involvement/wasting was noted in three MM patients but not in LGMD2B. A highly statistically significant finding was a more pronounced involvement of the lower limbs in comparison with the upper limbs (p=0.00007) in our patients. Comparison of upper and lower limb muscle groups revealed that power was decreased to 76% (±4%) in the upper limbs compared with 49% (±5%) in the lower limbs in our patient cohort enabling relatively preserved muscle function in the arms and hands. Five patients were wheelchair dependent and one patient required intermittent wheelchair use (table 3). Four of these patients initially presented with an LGMD2B and two patients with an MM phenotype.

{kind=link}
{kind=link}
{kind=link}
Muscle strength. The weakest muscle functions after median disease duration of 9 years were plantar flexion, hip adduction and hip extension.
Description of patient cohort
Pulmonary and cardiac function
Respiratory vital capacity was not reduced to a clinically relevant level in any of the patients nor did any of the patients report symptoms compatible with nocturnal hypoventilation. Although not formally assessed, none of the patients has presented with clinically overt cardiovascular symptoms so far, with the longest disease duration being 42 years.
Pre-symptomatic muscle function
As anecdotal evidence has previously suggested good muscle prowess before onset of symptoms in patients affected by dysferlin deficiency, a detailed history was taken in all patients on their sporting and physical activities and capabilities prior to onset of symptoms. Fifty-three per cent of dysferlin deficient patients reported a high degree of activity and were remarkably sporty. No statistically significant difference was noted regarding age of onset between the group of patients with marked sportiness in comparison with those who were average or not good at sports (p=0.77). It is difficult to provide a direct comparison with another diagnostic groups on this point. However, when we addressed the same question in a group of 22 LGMD2A patients, only 9% reported good muscle prowess before onset of symptoms. No statistically significant difference between MM and LGMD2B was found with respect to pre-symptomatic fitness levels, suggesting that this parameter is not associated with a specific phenotype of dysferlin deficiency at presentation.
Discussion
In this first UK study of patients affected by dysferlinopathy, we observed that a subgroup of patients had an earlier age of onset than previously reported with 25% of patients manifesting before the age of 13 years. No other factors were identified distinguishing this group from the rest of the patients (mutation, pathology data, levels of physical activity prior to onset). In addition, one previously reported patient showed first symptoms at the age of 73 years which is the oldest age of disease manifestation reported so far.26 Taken together, these data highlight a wider range of age of onset than previously recognised4 6 8 and stress the fact that dysferlinopathy needs to be considered as a differential diagnosis in children presenting with limb girdle weakness or a distal myopathy, and that muscular dystrophy should not be discounted in patients with advanced age. This broad range of age at onset is further supported by a recent report of two siblings with dysferlin deficiency presenting with congenital onset.27
More than half of our patients (53%) described a high degree of sportiness prior to onset of symptoms. This finding appears to be more common in dysferlin deficiency as this is not a feature regularly associated with any other form of muscular dystrophy known to date. Direct clinical comparison of different forms of muscular dystrophy is difficult and always represents a compromise as there is no direct match between age of onset, clinical presentation and severity. However, we compared our findings in dysferlin deficient patients to patients affected by LGMD2A. In contrast with patients affected by dysferlin deficiency, the majority of calpain deficient patients showed impairment of muscle function before the onset of overt weakness as they were significantly more often not good at sports/slow or avoided sports altogether before disease onset. The factor(s) responsible for this ‘sporty’ phenotype in dysferlin deficiency are unknown, but this finding suggests that these patients may not only have normal/preserved muscle function but may actually have very good muscle function prior to onset of wasting and weakness. Impaired sarcolemmal resealing of damaged myofibres has been identified as a key pathomechanism leading to muscular dystrophy in the dysferlin knockout mouse.12 But this mechanism cannot conclusively explain why dysferlin deficient patients exhibit good muscle prowess before onset of weakness and wasting. An exaggerated turnover of satellite cells at an early stage of the disease could potentially contribute to this phenotype but this possibility has not been investigated so far.
Although the same mutation may lead to initial presentation as MM or LGMD2B even within the same family,28 29 we did not observe this degree of intrafamilial variability in our cohort of patients.30 In contrast with a recent study,4 most of our patients did present with either proximal or distal onset and only one patient displayed proximal and distal involvement simultaneously, although with disease progression we noted as have others that it was difficult to distinguish those who had initially presented with proximal or distal involvement. The larger number of patients with proximal presentation in our study may also reflect a referral bias as our diagnostic service is primarily orientated towards the LGMDs. We did not identify any patient presenting with distal anterior involvement,3 similar to the observation by Nguyen et al,4 suggesting that this appears to be a rare presentation of dysferlinopathy.
The muscle most severely affected in our group of patients was the gastrocnemius muscle leading to an inability to stand on tiptoes also in patients initially presenting with a limb girdle phenotype. This has to take into account that the median disease duration at muscle assessment was 9 years (range 2.5–42 years). Overall weakness appears to be more pronounced in the lower than the upper limbs.
Two patients developed symptoms shortly after delivery of their first child, and one patient after delivery of her third child which to our knowledge has not been reported in dysferlin deficiency so far. This may suggest that pregnancy could possibly trigger onset of symptoms.
Mutations were spread along the entire gene, corroborating the findings that no mutational hot spots exist in the dysferlin gene (figure 1).7 In addition, our findings are in line with most previous reports in that we did not find any genotype–phenotype correlation either with regard to the nature of the mutation and severity or with the phenotype and age at presentation. In agreement with most data published so far but in contrast with a recently published study,11 we could not find a correlation between residual amounts of dysferlin on western blotting or immunohistochemistry and the resulting phenotype.
Recently, two patients have been reported as symptomatic dysferlin gene mutation carriers with heterozygous missense mutations in exons 18 and 20, respectively.31 Similarly, the brother of patient 12a who is affected by LGMD2B based on a compound heterozygous mutation in the dysferlin gene appears to be a symptomatic carrier with no second mutation detected (patient 12b). His mutated allele was maternally inherited (truncating mutation/deletion, exon 8). He presented later in life than his brother and symptoms were less severe with pain in his legs and prolonged recovery time after exercise without overt muscle weakness. His serum CK was slightly raised (300 IU, normal <130 IU), and the muscle biopsy showed myopathic features and patchy dysferlin labelling at the sarcolemma on immunohistochemistry similar to the two previously reported symptomatic carriers.31 Although dysferlin and calpain 3 appeared normal on western blot, it is highly likely that the clinical and biochemical findings are based on the single truncating mutation in exon 8 of the dysferlin gene in this patient.
Taken together, the data presented in this study extend our knowledge on the phenotype of dysferlinopathy as age of onset may be broader than previously appreciated reaching from childhood to late in life. Furthermore, good muscle prowess before onset of weakness appears a common finding in this form of muscle disease, understanding of which may contribute to the elucidation of the underlying pathogenetic mechanisms in dysferlinopathy.
Acknowledgments
We thank Dr Wolfram Kress, Institute of Human Genetics, University of Würzburg, Germany, for performing some of the sequencing of the dysferlin gene.
References
Footnotes
Funding This work was supported by grants from the Deutsche Forschungsgemeinschaft (KL 1868/1-1 and 1868/2-1 to LK) and by a grant provided by the Muscular Dystrophy Campaign. Dysferlin research at Newcastle is also supported by Action Medical Research, the Association Française contre les Myopathies and the Jain Foundation. VS, HL and LK are members of the German Muscular Dystrophy Network (MD-NET 01GM0601) funded by the German Ministry of Education and Research (BMBF, Bonn, Germany), http://www.md-net.org. MD-NET and the University of Newcastle are partners of TREAT-NMD (EC, 6th FP, proposal No 036825; http://www.treat-nmd.eu). The limb girdle muscular dystrophy service in Newcastle is funded by the National Commissioning Group (NCG) as part of the National Diagnostic and Advisory Service for rare neuromuscular diseases.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.