Article Text

Abstract
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive neurodegenerative disorder of the motor neurons in the motor cortex, brainstem and spinal cord. A combination of upper and lower motor neuron dysfunction comprises the clinical ALS phenotype. Although the ALS phenotype was first observed by Charcot over 100 years ago, the site of ALS onset and the pathophysiological mechanisms underlying the development of motor neuron degeneration remain to be elucidated. Transcranial magnetic stimulation (TMS) enables non-invasive assessment of the functional integrity of the motor cortex and its corticomotoneuronal projections. To date, TMS studies have established motor cortical and corticospinal dysfunction in ALS, with cortical hyperexcitability being an early feature in sporadic forms of ALS and preceding the clinical onset of familial ALS. Taken together, a central origin of ALS is supported by TMS studies, with an anterograde transsynaptic mechanism implicated in ALS pathogenesis. Of further relevance, TMS techniques reliably distinguish ALS from mimic disorders, despite a compatible peripheral disease burden, thereby suggesting a potential diagnostic utility of TMS in ALS. This review will focus on the mechanisms underlying the generation of TMS measures used in assessment of cortical excitability, the contribution of TMS in enhancing the understanding of ALS pathophysiology and the potential diagnostic utility of TMS techniques in ALS.
- ALS
- EMG
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
The term amyotrophic lateral sclerosis (ALS) was first coined by Charcot, who postulated the primacy of the upper motor neuron (UMN) in ALS pathogenesis.1 Assessment of cortical function in ALS and identification of the characteristic clinical phenotype involving combined upper and lower motor neuron abnormalities remain the key for ALS diagnosis.2–4 However, despite Charcot's initial observations, the site of disease onset and mechanisms underlying ALS pathophysiology remain areas of intense study and debate.5 In this setting, assessment of motor cortical and corticospinal function using non-invasive techniques, such as transcranial magnetic stimulation (TMS), has enhanced our understanding of ALS pathophysiology and resulted in novel diagnostic approaches.
Single-, paired- and multiple-pulse TMS techniques have all been used (figure 1) with the following measures taken to reflect corticomotoneuronal function: motor threshold (MT), motor evoked potential (MEP) amplitude, central motor conduction time (CMCT), cortical silent period (CSP), intracortical inhibition and facilitation. The present review will focus on the mechanisms underlying the generation of these TMS measures, while at the same time assessing the contributions TMS has made in the understanding of ALS pathophysiology. With an eye towards the future, the review will also consider the potential diagnostic utility of TMS in ALS and incorporation of TMS as a disease biomarker in the assessment of neuroprotective medications in a clinical trial setting.

Transcranial magnetic stimulation excites a network of neurons in the underlying motor cortex with motor evoked potentials recorded over the contralateral abductor pollicis brevis muscle. The motor cortex is preferentially stimulated when the current flows in a posterior–anterior direction within the motor cortex.
Background TMS terminology and pathophysiology
MT reflects the ease with which corticomotoneurons are excited and is proposed to be assessed by the International Federation of Clinical Neurophysiology as the minimum stimulus intensity required to elicit a small (usually >50 μV) MEP in the target muscle in 50% of trials.6 With the recent adaptation of threshold tracking techniques, MT can also be measured as the stimulus intensity required to elicit and maintain a target MEP response of 0.2 mV.7–9 MT reflects the density of corticomotoneuronal projections onto the spinal motor neuron with the highest density of projections to intrinsic hand muscles having the lowest MTs.10–12 MTs are lower in the dominant hand12 and correlate with the ability to perform fine (fractionated) finger tasks,13 so that MT has the potential to map corticomotoneuronal representation and function.
As well as reflecting the density of corticomotoneuronal projections, MTs may also be a biomarker of cortical neuronal membrane excitability.14–16 MTs are influenced by the glutamatergic neurotransmitter system, through α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) receptors, whereby excessive glutamate activity reduces MTs.17 In contrast, pharmacological blockade of voltage-gated sodium channels raises MT.18
In ALS, abnormalities in MT have been inconsistent. While some TMS studies reported an increased MT or even an inexcitable motor cortex,19–26 others have documented either normal or reduced MT.27–32 These discrepancies likely relate to heterogeneity of the ALS phenotype and the stage of disease at time of testing and rate of progression. Longitudinal studies have documented a reduction of MTs early in the disease course, increasing to the point of cortical inexcitability with disease progression.29 The early reduction in MT appears most pronounced in ALS patients with profuse fasciculations, preserved muscle bulk and hyper-reflexia.33 Fasciculations may precede other features of ALS by many months and taken in association with reduced MT suggest a cortical origin of fasciculations in ALS.34 Reduced MT may be modulated by increased glutamate excitation, reduced gamma-Aminobutyric acid (GABA) inhibition or a combination of both. Reduced MT early in ALS supports an anterograde transsynaptic process, whereby cortical hyperexcitability underlies the development of progressive neurodegeneration.
MEP amplitude reflects a summation of complex corticospinal volleys consisting of D (direct)- and I (indirect)-waves.14 ,35 At threshold, TMS elicits I-waves at intervals of 1.5 ms, which increase in amplitude with increasing stimulus intensity.35 The increase in MEP amplitude with increasing stimulus intensity may be used to generate a stimulus–response curve that follows a sigmoid function.36 As with MT, the MEP amplitude reflects the density of corticomotoneuronal projections onto motor neurons.37 When compared with MT, the MEPs probably assess the function of neurons that are less excitable or further away from the centre of the TMS induced electrical field.38 The MEP amplitude should be expressed as a percentage of the maximum compound muscle action potential (CMAP) evoked by electrical peripheral nerve stimulation.6 Doing so takes into account any lower motor neuron pathology and provides insight into the percentage of the motor neurone pool activated in the MEP. Normative values for the MEP to CMAP ratio demonstrate a large inter-subject variability thereby reducing the sensitivity and limiting the value of this measure for detecting abnormalities of the corticomotoneurons.38 ,39
The MEP responses are modulated by a variety of neurotransmitter systems within the central nervous system.37 ,40 Specifically, GABAergic neurotransmission via GABAA receptors suppresses while glutamatergic and noradrenergic neurotransmission enhances the MEP amplitude.41 Of interest, these changes in MEP amplitude occur independently of changes in MT, suggesting that physiological mechanisms underlying the generation of the MEP amplitude and MT are varied.
Abnormalities of MEPs have been extensively documented in ALS.38 Increases in MEP amplitude have been reported in sporadic and familial forms of ALS (figure 2A), most prominently early in the disease course.30 ,31 ,42 MEP amplitude correlates with surrogate biomarkers of axonal degeneration, such as the strength duration time constant, thereby providing an association between cortical hyperexcitability and motor neuron degeneration.30 ,43 The increase in MEP amplitude in ALS is not seen in mimic disorders despite a comparable degree of lower motor neuron dysfunction (figure 2B). This suggests that the MEP amplitude changes in ALS are excitotoxic in nature.44–47
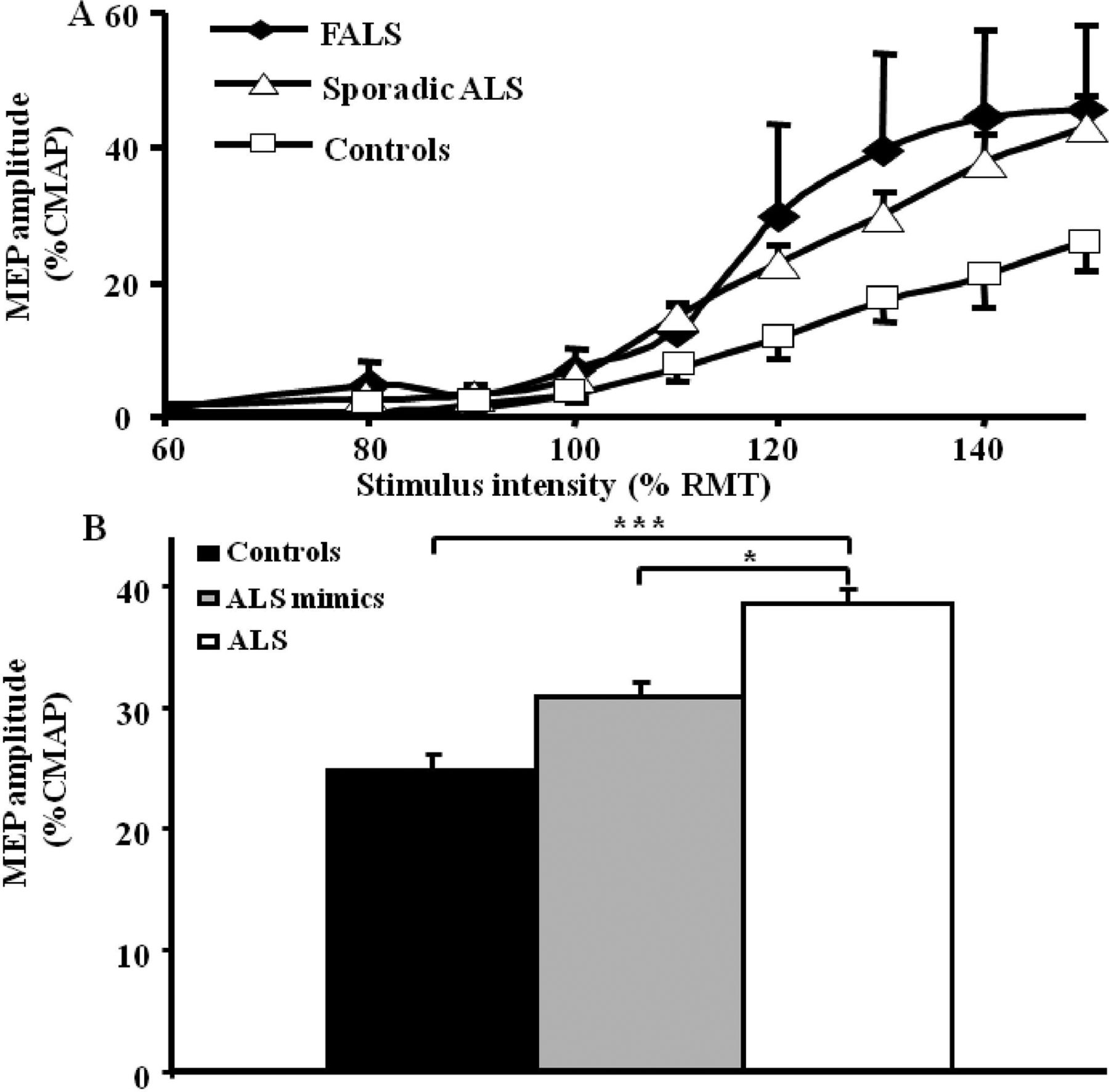
(A) The motor evoked potential (MEP) amplitude, expressed as a percentage of compound muscle action potential (CMAP) response, is significantly increased in sporadic amyotrophic lateral sclerosis (ALS) and familial ALS (FALS) when compared with healthy controls. (B) The MEP amplitude is significantly increased in ALS when compared with pathological and healthy controls, thereby distinguishing ALS from ALS mimic disorders.* p<0.05; ***p<0.001. RMT, resting motor threshold.
CMCT represents the time from stimulation of the motor cortex to the arrival of corticospinal volley at the spinal motor neuron.6 Multiple factors contribute to the CMCT including time to activate the corticospinal cells, conduction time of the descending volley down the corticospinal tract, synaptic transmission and activation of spinal motor neurons.48 CMCT may be measured using either the F-wave or cervical (or lumbar) nerve root stimulation methods;49 ,50 both methods provide only an estimation of the CMCT,48 ,51 and given that a variety of technical, physiological and pathological factors influence CMCT,48 there is a range of normative data.
In ALS, CMCT is typically modestly prolonged,21 ,29 ,52 probably reflecting axonal degeneration of the fastest conducting corticomotoneuronal fibres and increased desynchronisation of corticomotoneuronal volleys secondary to axonal loss.28 ,53 ,54 The D90A-SOD1 ALS mutation is a unique exception; in this disorder CMCT is typically very prolonged.55 The sensitivity of detecting a prolonged CMCT may be improved by recording from both upper and lower limb muscles, or from cranial muscles in ALS patients with bulbar onset disease.26 ,56
CSP refers to the interruption of voluntary electromyography activity in a target muscle induced by stimulation of the contralateral motor cortex.57 The CSP duration is measured from the onset of the MEP response to resumption of voluntary electromyography activity37 ,57 and increases with stimulus intensity.57–59
The CSP is mediated by both spinal mechanisms, in its early part, and cortical inhibitory neurons acting via GABAB receptors in the latter part.57 ,58 ,60–63 Since the duration is determined by the latter part, the CSP is a measure of cortical inhibition. In addition, the density of the corticomotoneuronal projections onto motor neurons also influences the CSP, with the CSP duration being the longest for upper limb muscles.38
Abnormalities of the CSP duration are well established in ALS.37 Absence or reduction in CSP duration has been reported in both sporadic and familial ALS, with the reduction of CSP duration being the most prominent early in the disease course.30–32 ,44 ,46 ,52 ,64–67 The reduction of CSP duration appears to be specific for ALS among neuromuscular disorders, being normal in X-linked bulbospinal muscular atrophy (Kennedy's disease), acquired neuromyotonia and distal hereditary motor neuronopathy with pyramidal features.44–47 Although the mechanisms underlying CSP duration reduction in ALS remain to be established, decreased motor drive and reduced GABAergic inhibition, either due to degeneration of inhibitory interneurons or dysfunction of GABAB receptors, may underlie the reduction of CSP duration in ALS.
An absent or delayed ipsilateral CSP has also been reported as an early abnormality in ALS.67 ,68 The ipsilateral CSP depends on functioning of transcallosal glutamatergic fibres projecting onto inhibitory interneurons in the non-stimulated motor cortex,69 and degeneration of these transcallosal fibres or their targeted inhibitory interneurons may account for abnormalities of the ipsilateral CSP in ALS.
Paired-pulse TMS techniques
The previous section has covered conventional TMS parameters that can be assessed through activation of the motor cortex by single impulses. Motor cortical excitability may also be assessed using paired-pulse techniques, in which a conditioning stimulus modulates the effect of a second test stimulus. Several different paired-pulse paradigms have been developed,37 ,38 but short interval intracortical inhibition (SICI), intracortical facilitation (ICF) and long interval intracortical inhibition have been most frequently used in ALS clinical research as methods to determine cortical excitability.
To identify SICI and ICF, a subthreshold conditioning stimulus is typically delivered at predetermined time intervals prior to a suprathreshold test stimulus.8 ,70–72 In the early TMS paradigms,70 ,72 ,73 the conditioning and test stimuli were kept constant, and changes in the test MEP amplitude were evaluated. Typically, if the interstimulus interval (ISI) was between 1 and 5 ms, the test response was inhibited (SICI). Increasing the ISI to between 7 and 30 ms resulted in the facilitation of the test response (ICF).38
By recording the descending corticospinal volleys through epidural electrodes at the level of the cervical spinal cord, it has been deduced that both SICI and ICF originate at the level of the motor cortex.35 ,72 Specifically, SICI is associated with a reduction in the number and amplitude of late I-waves, namely I2 and I3, with I-wave suppression remaining up to an ISI of 20 ms, which is the typical duration of the inhibitory postsynaptic potential mediated through GABAA receptors.71 ,74 SICI and ICF appear to be physiologically distinct processes as evident by lower thresholds for activation of SICI and SICI remains independent of the direction of current flow in the motor cortex induced by a subthreshold conditioning pulse in healthy subjects, while ICF appears to be preferentially generated by current flowing in a posterior–anterior direction.73
A limitation of the ‘constant stimulus’ paired-pulse technique has been the marked variability in MEP amplitudes with consecutive stimuli.71 ,75 To overcome this limitation, a threshold tracking technique was developed whereby a constant target MEP response (0.2 mV) was tracked by a test stimulus.7 ,8 Using threshold tracking, two phases of SICI were identified,7 ,8 ,76 ,77 a smaller phase at ISI ≤1 ms and a larger phase at ISI 3 ms (figure 3A). Although synaptic neurotransmission through the GABAA receptor mediates the second phase of SICI,74 ,78–80 the precise mechanisms underlying the first phase of SICI remain uncertain. It was initially suggested that the first phase of SICI reflected local excitability properties, particularly relative refractoriness of cortical axons, with resultant resynchronisation of cortico-cortical and corticomotoneuronal volleys.7 ,81 Subsequently, it has been argued that synaptic processes best explain the development of the initial phase of SICI, possibly driven by activation of cortical inhibitory circuits that were distinct to those that mediated the later SICI phase.76 ,77 ,82

(A) Short interval intracortical inhibition (SICI), defined as the stimulus intensity required to maintain a target motor evoked potential of 0.2 mV, as assessed by the threshold tracking transcranial magnetic stimulation technique. Intracortical inhibition is illustrated by an increase in the conditioned test stimulus intensity required to track the target response, while intracortical facilitation is indicated by a reduction in test stimulus intensity. In healthy controls, SICI develops between interstimulus intervals (ISI) of 1 and 7 ms, with two peaks evident at 1 and 3 ms as indicated by the arrows. Intracortical facilitation developed between ISIs of 10 and 30 ms. SICI is significantly reduced in both sporadic amyotrophic lateral sclerosis (SALS) and familial amyotrophic lateral sclerosis (FALS). (B) Averaged SICI, between ISI 1 and 7 ms, was reduced in two presymptomatic superoxide dismutase-1 (SOD-1) mutation carriers 6 months prior to the development of ALS. (C) Normalised SICI, expressed as a fraction of the SICI value measured at the first study, was reduced 8 months prior to development of ALS in a third presymptomatic SOD-1 mutation carrier.
A reduction or absence of SICI, together with an increase in ICF, indicative of cortical hyperexcitability has been documented in cohorts of sporadic and familial ALS patients (figure 3A).30–32 ,44 ,45 ,83–88 Of relevance, cortical hyperexcitability appears to be an early feature in sporadic ALS, correlating with measures of subsequent peripheral neurodegeneration.30 In addition, cortical hyperexcitability appeared as an early feature in familial ALS due to mutations linked to the superoxide dismutase-1 (figure 3A) and fused in sarcoma (FUS) genes,31 preceding the clinical development of familial ALS (figure 3B).31
Neuropathological studies in ALS have identified degeneration of inhibitory cortical interneurons89 and this could account for the reduction in SICI. Separately, glutamate-mediated excitotoxicity may also contribute to SICI reduction, as was suggested by partial correction of SICI abnormalities in ALS patients treated with the glutamate antagonist riluzole.87 A recent study documenting SICI reduction at low (40% of resting MT (RMT)), medium (70% of RMT) and high (90% of RMT) conditioning stimulus intensities in ALS patients provided further support for the notion that abnormalities in SICI appeared to be mediated by a combination of glutamate excitotoxicity and degeneration of inhibitory cortical circuits.90 As such, preserving the integrity of intracortical inhibitory circuits, and counteracting excitatory cortical circuits, may serve as potential therapeutic options in ALS.
Utility of peristimulus time histograms
A peristimulus time histogram technique can assess the function of a select subset of corticomotoneurons by recording the perturbation of voluntarily recruited motor units induced by a threshold cortical stimulation.53 In healthy controls, there is a well synchronised primary peak with a latency of approximately 20–30 ms recording from hand or forearm muscles.28 ,53 Analysis of this primary peak in disease states such as ALS provides information on corticomotoneuronal conduction time, the extent of desynchronisation of corticomotoneuronal descending volleys, the degree of corticomotoneuronal synaptic input onto the anterior horn cell and the timing of excitatory and inhibitory inputs to the motor neuron.33
In ALS, the primary peak becomes desynchronised, prolonged in duration and delayed.28 ,91 ,92 In addition, the amplitude of the primary peak may be increased with additional subcomponents both suggestive of corticomotoneuronal hyperexcitability.53 ,93 These primary peak abnormalities appear early in ALS, accompanied by reduced MTs. With progression of disease, there is prolongation and increased desynchronisation of the primary peak, findings possibly specific to ALS when compared with healthy controls and Kennedy's disease.53 ,94
Triple stimulation technique
Over recent years, collision techniques such as the triple stimulation technique (TST) have been used to reduced the degree of MEP desynchronisation which normally occurs following a single cortical stimulus.95 ,96 This complex technique is performed by first delivering a high-intensity magnetic stimulus to motor cortex followed by supramaximal electrical stimulation of the peripheral nerve supplying the target muscle at the wrist such that the descending corticomotoneuronal volley is ‘collided’ out by the antidromic action potentials. Collision takes place along the proximal segment of the peripheral nerve at the upper arm. A third stimulus is subsequently delivered to Erb's point (axilla) after an appropriate delay, eliciting a highly synchronised motor response in those fibres in which the collision had occurred. The amplitude and area of this test CMAP response are compared with the response induced by the conditioned TST paradigm (Erb's point-wrist–Erb's point stimulation) yielding an amplitude ratio of >93% and area ratio of >92% in healthy controls.95 ,96
In ALS, the TST is sensitive at detecting subclinical corticomotoneuronal dysfunction.54 ,97 Corticomotoneuronal dysfunction was also reported in Kennedy's disease using the TST technique,98 ,99 potentially limiting the diagnostic utility of TST in ALS. Recently, however, a combination of TST with single- and paired-pulse TMS techniques has reaffirmed the functional integrity of corticomotoneuronal tracts in Kennedy's disease,100 and thereby the diagnostic utility of TST.
Diagnostic biomarker in ALS
Given the well documented TMS abnormalities in ALS patients, the TMS techniques may be of utility in the diagnostic process of ALS. Although UMN signs may be clinically evident in ALS, in some phenotypes such as the flail arm variant, this may not be the case, and detection of subclinical UMN dysfunction may facilitate the diagnosis.42 Abnormalities of cortical excitability, including an increase in MEP amplitude along with reduction of SICI and RMTs, have been reported in the flail-arm variant of ALS, underscoring the utility of TMS in detecting subclinical UMN dysfunction.42 Of further relevance, subclinical UMN dysfunction has been reported in progressive muscular atrophy (PMA),101–103 suggesting that PMA may be a phenotype of ALS. While corticomotoneuronal integrity was recently reported to be intact in PMA using a β-band intermuscular coherence technique,104 assessment of cortical function with TMS techniques may be of diagnostic utility, especially in light of presence of subclinical UMN pathology in PMA.102 ,103
Importantly, single- and paired-pulse TMS techniques reliably distinguish ALS from the mimic disorders (table 1), hastening the diagnosis of ALS by up to 8 months.47 A reduction in averaged SICI, between ISI 1 and 7 ms, and peak SICI at ISI 3 ms were the most robust diagnostic TMS parameters, with the finding of absent SICI exhibiting a sensitivity of 97%.47 Of further relevance, TMS studies have established the presence of early and subclinical dysfunction of cortico-bulbar and cortico-respiratory tracts in ALS,26 ,105–107 thereby suggesting a potential diagnostic utility of bulbar and diaphragmatic MEP recordings. In addition, combining TMS with radiological techniques, such as MR spectroscopy, may further add to the diagnostic yield especially given the sensitivity of MR spectroscopy in detecting subclinical UMN dysfunction.108–110 Consequently, combining TMS techniques, in particular the recording of SICI as well as bulbar and diaphragmatic MEPs, together with radiological techniques, such as MR spectroscopy, may enable an earlier diagnosis of ALS and thereby commencement of neuroprotective therapies and recruitment into clinical trials.
Transcranial magnetic stimulation (TMS) techniques in amyotrophic lateral sclerosis (ALS) mimic disorders
In addition to its diagnostic utility, it has been suggested that TMS may exhibit a clinical utility in assessing disease progression in ALS.111 Specifically, longitudinal TMS studies in ALS patients reported a significant reduction in MEP amplitude, MT and CMCT, and suggested that reduction in MEP amplitude may be an objective biomarker of disease progression in ALS.111 In contrast, others have failed to document any significant longitudinal changes in TMS parameters, thereby arguing against TMS utility in the monitoring of disease progression in ALS.52 Prospective longitudinal studies are indicated to further clarify the role for TMS in monitoring disease progression in ALS.
Concepts of ALS pathophysiology
In his original writings, Charcot concluded that ALS was a disorder of the brain and that the lower motor neuron component resulted from a downstream affect. Not all his contemporaries agreed and in particular Gowers was adamant that the demise of upper and lower motor neurons were independent events. In the past 2 decades the site of ALS onset has been revisited, to a large extent precipitated by the advent of TMS. Three schools of thought have developed pertaining to the role of the UMN, and related pathophysiological processes in ALS: (i) ‘the dying forward’ hypothesis, (ii) ‘the dying back’ hypothesis and (iii) ‘the independent degeneration’ hypothesis (figure 4). While the site of disease onset in ALS remains uncertain, TMS studies have tended to favour a cortical origin, with excitotoxicity mediating motor neuron degeneration in ALS.5 ,112

{kind=link}
{kind=link}
{kind=link}
{kind=link}
The dying forward and dying back hypothesis of amyotrophic lateral sclerosis (ALS). The ‘dying forward’ hypothesis proposed that ALS was primarily a disorder of the corticomotoneurons (highlighted in red), with anterior horn cell degeneration mediated via an anterograde glutamate-mediated excitotoxic process. In contrast, the dying back hypothesis proposes that ALS begins within the muscle or neuromuscular junction, with pathogens retrogradely transported from the neuromuscular junction to the cell body where these pathogens may exert their deleterious effects.
The dying forward hypothesis proposes that ALS is primarily a disorder of the corticomotoneurons, which connect monosynaptically with anterior horn cells.113 Corticomotoneuronal hyperexcitability was postulated to induce anterior horn cell degeneration transsynaptically via an anterograde glutamate-mediated excitotoxic process.113 Most TMS studies have demonstrated that cortical hyperexcitability is an early feature in sporadic and familial ALS, linked to motor neuron degeneration.27 ,30 ,31 ,43 ,65 ,112 ,114 ,115 In addition, longitudinal studies in asymptomatic SOD-1 mutation carriers revealed that cortical hyperexcitability developed prior to the clinical onset of ALS,31 also seen in the G93A SOD-1 mouse model.116 Of relevance, loss of parvalbumin-positive inhibitory interneurons in the motor cortex of ALS patients would contribute to the development of cortical hyperexcitability.117 In keeping with a cortical origin of ALS is the now accepted view that ALS and frontotemporal dementia (FTD) represent an overlapping continuum of the same disorder,118 ,119 an observation underscored by recent genetic findings establishing that increased hexanucleotide repeat expansion in the first intron of C9ORF72 gene on chromosome 9p21 is associated with both ALS and FTD.120 ,121 Of further relevance, accumulation of TDP-43 ubiquitinated inclusions in anterior horn cells appears to be a pathological hallmark of ALS.119 ,122 Interestingly, identical TDP-43 inclusions may also be evident in cortical neurons within the frontal (Betz cells) and temporal lobes of ALS patients,119 ,122 ,123 underscoring the link between FTD and ALS, and thereby a cortical origin of ALS.
Of relevance, molecular approaches have provided further corroborating evidence for glutamate excitotoxicity in ALS. Specifically, a significant reduction in the expression and function of the astrocytic glutamate transporter, excitatory amino acid transporter 2 (EAAT2), has been reported in the SOD-1 mouse model and the motor cortex and spinal cord of ALS patients.124–128 In addition, dysfunction of EAAT2 transporter appeared to be a preclinical feature in the SOD-1 mouse model.129 ,130 Further underscoring the importance of astrocytes in ALS pathophysiology are recent stem cell studies documenting that motor neuron degeneration appears to be initiated by dysfunction of astrocytes.131
On the postsynaptic side, increased expression of glutamate receptors permeable to excessive influx of Na+ and Ca2+ ions132 have been reported in ALS,133–137 potentially rendering the motor neurons more susceptible to glutamate excitotoxicity.138 Further support for a role for glutamate excitotoxicity has been indirectly provided by the clinical benefit of riluzole, a glutamate antagonist, in ALS patients.139–142
For the glutamate hypothesis to be a plausible mechanism of motor neuron degeneration, the issue of selectivity of motor neuron involvement in ALS, together with sparing of motor neurons in non-ALS conditions exhibiting cortical hyperexcitability,38 must be explained. A number of molecular features may render the motor neurons vulnerable to glutamate toxicity in ALS. First, motor neurons preferentially express glutamate receptors, such as the AMPA receptors, which are more permeable to influx of Ca2+ ions.133 ,134 ,136 ,137 In addition, motor neurons in ALS patients lack the intracellular expression of Ca2+ binding proteins parvalbumin and calbindin D28k, both required to buffer intracellular Ca2+.143 ,144 Aberrant activity of the inositol 1,4,5-triphosphate receptor type 2 receptor has been reported in ALS,145 ,146 thereby resulting in higher intracellular concentrations of Ca2+ within the motor neurons. Ultimately, an influx of Ca2+ ions through the ionotropic glutamate receptors NMDA occurs in the motor neurons,147 ,148 resulting in increased intracellular Ca2+ concentration and activation of Ca2+-dependent enzymatic pathways that mediate neuronal death.149–151 Glutamate excitotoxicity may also result in production of free radicals that can further damage intracellular organelles and thereby cause cell death.152–154
It could be argued that the finding of widespread fasciculations in ALS, an important diagnostic criterion,155 may argue against a dying forward mechanism given that fasciculations are thought to originate from the distal motor axon, are associated with abnormalities of sodium and potassium conductance, and may precede the onset of lower motor neuron dysfunction.156–162 It seems unlikely that cortical hyperexcitability could lead to changes in distal axonal excitability that would result in widespread fasciculations. Importantly, a supraspinal mechanism for triggering fasciculations in ALS has been previously reported.34 In agreement with this notion are findings that fasciculations in ALS may originate at the level of the motor neuron cell body.157 As such, it could be hypothesised that hyperexcitability of descending motor pathways may contribute to generation of fasciculation in ALS, thereby providing additional support for a dying forward process.
In conjunction with glutamate excitotoxicity, there is compelling evidence that mitochondrial dysfunction may exert an important role in the pathophysiology of ALS.163–168 Under conditions of excessive Ca2+ load, as may be evident with glutamate excitotoxicity,169 mitochondrial production of free radicals increases resulting in injury of critical neuronal cellular proteins and DNA. Mitochondrial dysfunction may in turn enhance glutamate excitotoxicity by disrupting the normal resting membrane potential, resulting in loss of the voltage-dependent Mg2+-mediated block of NMDA receptor channels.170 From a therapeutic perspective, dexpramipexole, a pharmacological agent that enhances mitochondrial function,171 was effective in slowing ALS progression in a recent phase II trial.172 A phase III, multicentre, international trial was commenced in March 2011 to determine the clinical efficacy of dexpramipexole in ALS (ClinicalTrials.gov-NCT01281189). Taken further, it is anticipated that TMS studies will be used to determine the efficacy of dexpramipexole in the modulation of cortical excitability in an attempt to provide further insight into ALS pathophysiology.
The dying back hypothesis proposes that ALS is primarily a disorder of the lower motor neurons, with pathogens retrogradely transported from the neuromuscular junction to the cell body where they exert their deleterious effects.173 Although some pathological studies have indirectly supported a dying back process,174–176 no pathogens of any type have been identified in relation to ALS. The presence of widespread dysfunction within the frontal cortex, including the primary, supplementary and prefrontal motor cortices in ALS, remains difficult to reconcile with a dying back process.3 ,110 ,177 In addition, the absence of central pathology in other lower motor neuron disorders such as Kennedy's disease or poliomyelitis provides a further argument against a dying back process.33 ,44
The independent degeneration hypothesis suggests that the upper and lower motor neurons degenerate independently.178 Some 100 years after the original Gowers publication, neuropathological studies provided support for the independent degeneration hypothesis whereby the degeneration of upper and lower motor neurons appeared to be independent.179 ,180 These correlative morphological techniques, however, may be confounded by the anatomical and functional complexity of the corticomotoneuronal system.181 In particular, there remains considerable variability in the corticomotoneuronal to anterior horn cell ratio, due to synaptic changes, and as such attempts to correlate upper and lower motor neurons on autopsy studies may not be meaningful.33
In addition to the three competing theories of ALS pathogenesis, a prion-like propagation hypothesis has also been suggested.182 Specifically, the previously documented contiguous spread of ALS5 ,183 could be explained by direct neuron-to-neuron transmission of pathogenic proteins via exosomes, defined as small lipid membranous microvesicles.182 The pathogenic exosomes could spread in either a rostral direction, explaining the rostral-to-caudal spread of ALS, or in a lateral–horizontal direction accounting for the lateral-to-medial spread of disease. In addition, non-contiguous propagation of ALS could also be explained by spread of pathogenic proteins or toxic molecules through the blood or CSF via exosomes.182 Interestingly, the genes implicated in ALS pathogenesis, including TDP-43 and FUS, possess a putative prion domain.184 Although a prion-like propagation mechanism may seem an attractive explanation for the spread of ALS, at present there is no direct evidence to support such a process in ALS.
Future clinical utility of TMS
Although first described by Charcot some 150 years ago, the pathophysiological mechanisms underlying ALS, variability, rate of progression and site of disease onset remain obscure. Objective assessment of UMN function in ALS remains a difficult task in clinical neurophysiology.185 While TMS is mainly used as a clinical research tool, conducted in specialised neurophysiological laboratories, there is an urgent need to objectively assess UMN function in ALS. This has been underscored by the recent Awaji diagnostic criteria.155 ,186 Although needle electromyography is used by the criteria to objectively assess lower motor neuron dysfunction, the detection of UMN involvement is based solely on clinical examination. Much has recently been learnt about ALS from MRI, especially diffusion tensor MRI, functional imaging and network analysis,110 ,187–198 but these tools remain prohibitively expensive, not readily available and may exhibit a modest diagnostic sensitivity.190 Commercially available TMS systems that will enable an objective assessment of UMN function could be readily developed, facilitating the diagnosis of ALS. Such TMS systems may result in the development of more functional ALS biomarkers that could be used in future drug trials for early patient recruitment and monitoring of drug efficacy.
Acknowledgments
Funding support from the Motor Neuron Disease Research Institute of Australia (MNDRIA), Sylvia and Charles Viertel Charitable Foundation Clinical Investigator grant, Ramaciotti Foundation and National Health and Medical Research Council of Australia (Project grant numbers 510233 and 1024915) is gratefully acknowledged.
References
Footnotes
-
Contributors SV was involved in the design, research and writing of the review. UZ, AE, MH and MK were involved in the design, critiquing and editing of the review.
-
Funding Support from the Motor Neuron Disease Research Institute of Australia (MNDRIA), Sylvia and Charles Viertel Charitable Foundation Clinical Investigator grant, Ramaciotti Foundation and National Health and Medical Research Council of Australia (Project grant numbers 510233 and 1024915) is gratefully acknowledged.
-
Competing interests None.
-
Ethics approval Local ethics committee.
-
Provenance and peer review Commissioned; externally peer reviewed.