Article Text

Abstract
Objective and methods Dysferlin encoded by DYSF deficiency leads to two main phenotypes, limb girdle muscular dystrophy (LGMD) 2B and Miyoshi myopathy. To reveal in detail the mutational and clinical features of LGMD2B in Japan, we observed 40 Japanese patients in 36 families with LGMD2B in whom dysferlin mutations were confirmed.
Results and conclusions Three mutations (c.1566C>G, c.2997G>T and c.4497delT) were relatively more prevalent. The c.2997G>T mutation was associated with late onset, proximal dominant forms of dysferlinopathy, a high probability that muscle weakness started in an upper limb and lower serum creatine kinase (CK) levels. The clinical features of LGMD2B are as follows: (1) onset in the late teens or early adulthood, except patients homozygous for the c.2997G>T mutation; (2) lower limb weakness at onset; (3) distal change of lower limbs on muscle CT at an early stage; (4) impairment of lumbar erector spinal muscles on muscle CT at an early stage; (5) predominant involvement of proximal upper limbs; (6) preservation of function of the hands at late stage; (7) preservation of strength in neck muscles at late stage; (8) lack of facial weakness or dysphagia; (9) avoidance of scoliosis; (10) hyper-Ckaemia; (11) preservation of cardiac function; and (12) a tendency for respiratory function to decline with disease duration. It is important that the late onset phenotype is found with prevalent mutations.
- Muscular Dystrophy
- Neurogenetics
- Clinical Neurology
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Dysferlinopathies are autosomal recessive muscular dystrophies caused by mutations in the dysferlin gene (DYSF, MIM# 603009). Dysferlin deficiency leads to two main phenotypes: limb girdle muscular dystrophy (LGMD) 2B and Miyoshi myopathy (MM).1 ,2 Dysferlin is located on the plasma membrane of skeletal muscle and is deficient in patients with MM and LGMD2B.3 ,4 However, atypical immunostaining in muscle from patients with dysferlin mutations occurs,5 ,6 and dysferlin expression is not normal in sarcoglycanopathy, dystrophinopathy,7 caveolinopathy8 ,9 or calpainopathy6 muscles. Therefore, the final diagnosis of dysferlinopathy requires identification of mutations in the dysferlin gene. We first reported dysferlin mutations in Japanese patients with MM10 and in a patient from a non-European ethnic group with distal anterior compartment myopathy (DACM),11 a relatively new phenotype of dysferlinopathy.12 Furthermore, we revealed that, in MM, four mutations (c.1566C>G, c.2997G>T, c.3373delG and c.4497delT) were relatively more prevalent in the Japanese population and the c.2997G>T mutation was associated with late onset.13 Although mutation analysis of the dysferlin gene is a time consuming task because of the large size of the gene,14 large series of patients with dysferlin gene mutations have been studied.6 ,15–24 However, few detailed analyses of the clinical features of LGMD2B, especially in relation to various types of mutations, have been reported. Here we report the clinical features of a series of 40 patients in 36 families with LGMD2B in whom dysferlin mutations were confirmed, and cardiac and respiratory functions were involved. In particular, we took into account the duration that had elapsed since onset when the clinical data were examined.
Materials and methods
We retrospectively observed 40 Japanese patients in 36 families with LGMD2B in whom dysferlin mutations were confirmed. LGMD was defined as symptomatic myopathy excluding MM and DACM at the first visit to a neurologist. Mutational analysis was performed by single strand conformation polymorphism analysis and sequencing on genomic DNA using our previously reported method,13 ,14 ,25 with minor modifications (Ref Seq NM_003494.2), and with informed consent and approval of our local ethics committee. We retrospectively reassessed the history of onset and progression of the disease. The clinical examination included manual muscle testing using the Medical Research Council (MRC) Scale and assignment of scales for the proximal limb muscles, as proposed by Brooke et al.26 Clinical examination was carried out by neurologists from the study groups for muscular dystrophy in Japan. Most patients had undergone muscle CT scans at some stage of the disease. Serum creatine kinase (CK) activity was measured. Cardiac and respiratory functions were evaluated. We selected the first and last manual muscle testing data and the last data on the scales for the proximal limb muscles, CK activity, cardiac function and respiratory function. We used all muscle CT scans. Patients were divided into three groups according to whether they had the homozygous c.2997G>T (p.Trp999Cys) mutation, the heterozygous c.2997G>T (p.Trp999Cys) mutation or other mutations. Difference in age at onset among the groups was evaluated by the Kruskal–Wallis test. Multiple comparison of age at onset between each group was assessed by Scheffé's F test. Difference in the first symptom among the groups was evaluated by the χ2 for independence test. Kaplan–Meier curves with log rank test were used to examine survival at each milestone of progression in the groups. Pearson's correlation coefficient test was used to identify significant associations in ejection fraction (EF) (n=21), atrial natriuretic peptide (n=9), brain natriuretic peptide (n=7), per cent vital capacity (%VC) (n=23), carbon dioxide partial pressure (pCO2) (n=16) and oxygen partial pressure (pO2) (n=16) with disease duration. Because supine and standing position chest x-rays were mingled, Pearson's correlation coefficient test was not used for the cardiothoracic ratio (n=22).
Results
Mutations
We identified 17 different mutations in 36 families (table 1). Two mutations (c.2974T>C (p.Trp992Arg) and c.2997G>T (p.Trp999Cys)) were missense mutations and four mutations (c.342+1G>A, c.937+1G>A, c.2643+1G>A and c.4794+1G>A) were splice site mutations. The others were nonsense mutations. The c.2997G>T (p.Trp999Cys) mutation was present in 26 alleles (36.1%). The c.1566C>G (p.Tyr522X) mutation and the c.4497delT mutation were both present in eight alleles (11.1%). We identified the c.3373delG mutation that has high frequency in Japanese patients with MM13 in only one allele.
Summary of dysferlin gene mutations of patients in this study
Clinical course
Mean age at onset of all patients was 26.6±9.9 years (range 14–58): the group homozygous for the c.2997G>T (p.Trp999Cys) mutation, 37.9±10.2 years (19–58); the group heterozygous for the c.2997G>T (p.Trp999Cys) mutation, 26.8±7.6 years (14–35); and the group without the c.2997G>T (p.Trp999Cys) mutation, 21.8±5.8 years (14–41). The difference between the group homozygous for the c.2997G>T (p.Trp999Cys) mutation and the group heterozygous for the c.2997G>T (p.Trp999Cys) mutation was significant (p<0.05). The difference between the group homozygous for the c.2997G>T (p.Trp999Cys) mutation and the group without the c.2997G>T (p.Trp999Cys) mutation was significant (p<0.01) (figure 1). The first symptom in most patients was lower limb weakness. Walking on tiptoe was the first sign in one patient. In three patients with the homozygous c.2997G>T (p.Trp999Cys) mutation, the first symptom was upper limb weakness. In two patients, one of them carrying the homozygous c.2997G>T (p.Trp999Cys) mutation, upper limb weakness was concomitant with lower limb weakness. There was a significant difference (p<0.01) in the probability that the first symptom was upper limb weakness among these three groups. The first symptom in one patient was left brachial pain and in another patient it was limitation of the elbow, hip, knee and spine.27 In the early stage of the clinical course, hypertrophy of the calves was noticed in three patients. Winged scapula, rigid spine27 and hollow foot were observed in one patient each. Lordosis was observed in two patients. Weakness of the face, dysphagia, scoliosis and kyphosis were not found. Choreic movements and pollakisuria were found in one patient.28 Mean duration at difficulty in running was 1.3 years from disease onset (range 0–10), 3.1 years (0–14) for difficulty in climbing stairs, 5.5 years (0–16) for stumbling, 6.0 years (0–10) for difficulty in standing on tiptoe, 6.7 years (0–15) for rising from the floor, 8.7 years (0–25) for noticing weakness in a proximal upper limb, 12.5 years (3–23) for walking with a cane, 20.4 years (9–35) for noticing weakness in a distal upper limb, 21.6 years (13–29) for using a wheelchair and 31.0 years (16–45) for using an electric wheelchair. There was a significant difference (p<0.05) in the survival ratio only at the stage of difficulty in standing on tiptoe among these three groups (figure 2). There were no significant differences in survival at the stage of the other symptoms between these three groups. According to the scales for the proximal limb muscles, only two patients each reached the severest stage of arms and shoulders (cannot raise hands to mouth and have no useful function) and hips and legs (confined to bed).

Histogram by age at onset. (A) Patients homozygous for the c.2997G>T mutation. (B) Patients heterozygous for the c.2997G>T mutation. (C) Patients without the c.2997G>T mutation.

Standing on tiptoe—possible survival curve. Survival curve of patients homozygous for the c.2997G>T mutation (line with squares), those heterozygous for the c.2997G>T mutation (line with triangles) and those without the c.2997G>T mutations (line with diamonds).
Manual muscle testing
In the first decade of the disease, the muscles of the lower limbs were predominantly involved. In the upper limbs, the deltoid muscle was predominantly involved. In the second decade of the disease, the biceps and triceps brachii muscles became weak. Later on, the flexor and extensor of the hand became weak. Muscle weakness progressed and, in the fifth decade of the disease, the muscles of the lower limbs were very weak (1 on the MRC Scale). In the patient with 52 years of disease duration, although the muscles of the upper and the lower limbs were 0 on the MRC scale, the flexor and extensor of the neck were relatively preserved (2 on the MRC Scale).
Muscle CT scans
The CT scans revealed low density changes in the gastrocnemius, especially in the medial heads, soleus, hamstrings and the erector spinal group, which was mainly affected in the lateral parts in the early disease stage. Low density abnormalities in the quadriceps femoris and the adductor magnus muscles were observed at close to 10 years after disease onset. The tibialis anterior, the peroneal group, the gluteal group, the quadrates lumborum and the deltoid muscles were involved at 10 years after onset. The gracilis and sarcorius muscles were preserved and had hypertrophied during the second decade of disease duration. Approximately 20 years after disease onset, low density changes in the dorsal muscles occurred in the neck region, particularly in the transversospinal group, except for the semispinalis capitis muscle. All muscles were replaced by low density tissue or were atrophied at 30 years after disease onset. The gluteal group, the psoas major muscle, the lateral abdominal group and the levator scapulae muscle were preserved in the late stage. There were exceptional cases. In one patient, low density changes in the dorsal muscles in the neck region occurred at an early stage. In another patient, the quadriceps femoris muscles were more severely damaged than the hamstrings at 14 years of disease duration.
Serum CK levels
Although serum CK levels were very high, there was a tendency for levels to decrease with disease duration (figure 3). There was a trend in the distribution of the levels by c.2997G>T (p.Trp999Cys) mutation. Levels in the group homozygous for the c.2997G>T (p.Trp999Cys) mutation were lower than those of the other two groups.
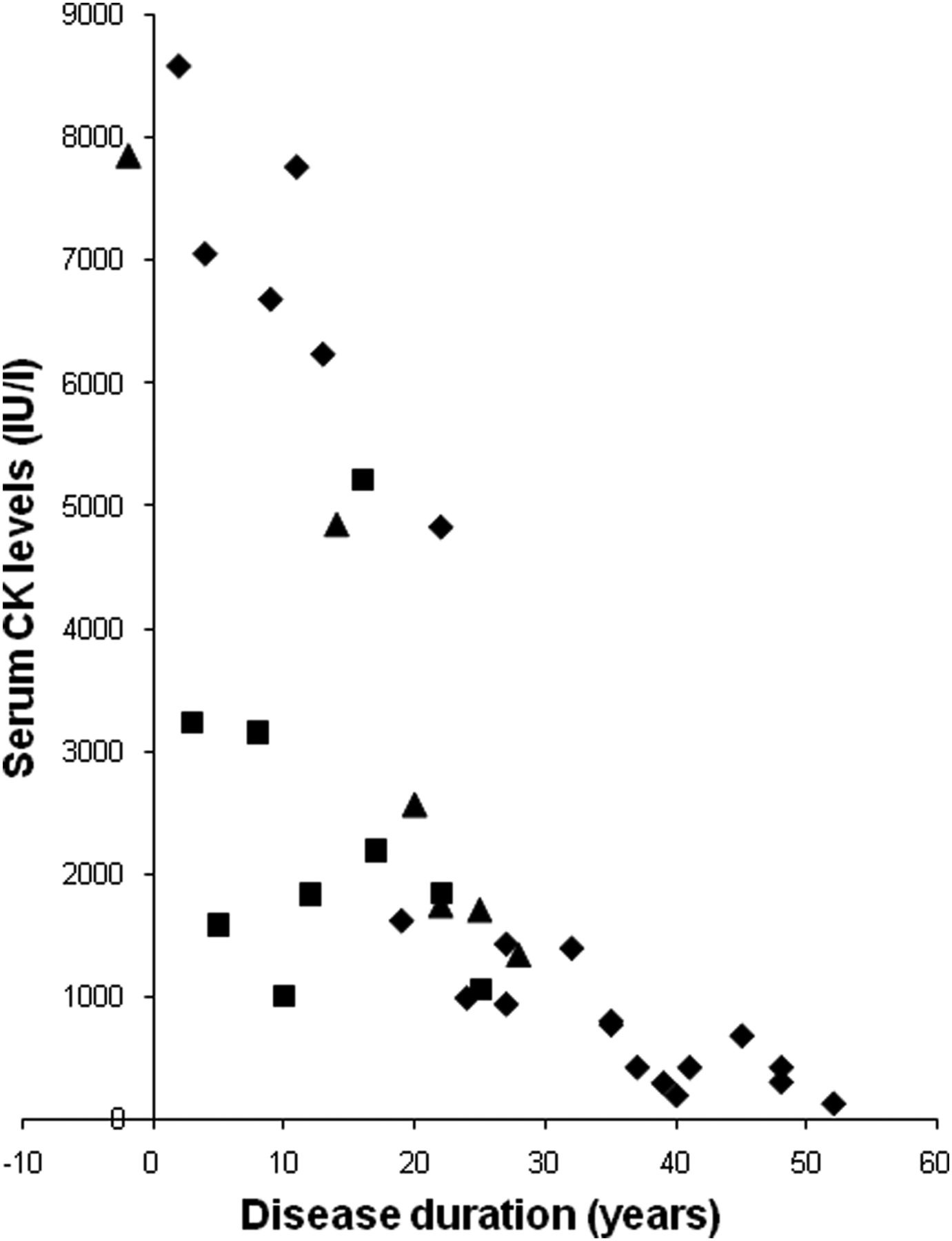
Serum creatine kinase (CK) levels during the disease course. Levels of serum CK in patients homozygous for the c.2997G>T mutation (squares), heterozygous for the c.2997G>T mutation (triangles) and those without the c.2997G>T mutations (diamonds).
Cardiac function
Although supine and standing position chest x-rays were mingled, in half of the patients the cardiothoracic ratio was >50%. No significant correlation was observed between EF and disease duration. Except for two patients, levels of EF were >50%. In all the patients, concentrations of atrial natriuretic peptide were within the normal range. Except for one patient, concentrations of brain natriuretic peptide were within the normal range. No significant correlation was observed between these peptides and disease duration. Although 19 patients exhibited normal findings on ECG, abnormalities were found in nine patients. The ECG showed premature ventricular contraction in one patient, one degree atrioventricular block in two patients, incomplete right bundle branch block in two patients, left axis deviation in two patients, left atrial hypertrophy in one patient, right ventricular hypertrophy in three patients, left ventricular hypertrophy in three patients, ST change in two patients and negative T in one patient.
Respiratory function
A statistically significant (p<0.01) correlation (r=−0.545) was observed between %VC and disease duration (figure 4). In 48% of patients, %VC was <80%. Although no significant correlation was observed between pCO2 and disease duration, in 44% of patients, levels of pCO2 were >45 mm Hg. In most patients, levels of pO2 were >70 mm Hg. No significant correlation was observed between pO2 and disease duration. Four patients had used non-invasive positive pressure ventilation (NIPPV). One patient who used NIPPV died after 42 years of disease duration from respiratory failure.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Respiratory function. (A) Per cent vital capacity (%VC) according to disease duration. The line is the regression line of %VC and disease duration. (B) Carbon dioxide partial pressure (pCO2) according to disease duration. (C) Oxygen partial pressure (pO2) according to disease duration. The triangles indicate levels in patients who had used non-invasive positive pressure ventilation. The cross (+) indicates the level in a patient who died after 42 years of disease duration of respiratory failure.
Discussion
Three mutations (c.2997G>T (p.Trp999Cys), c.1566C>G (p.Tyr522X) and c.4497delT) were relatively more prevalent in the present Japanese patients with LGMD2B. Also, in Japanese patients with MM, these three mutations were relatively more prevalent in our previous study.13 We identified the c.3373delG mutation that occurred with high frequency in Japanese patients with MM, as well as these three mutations,13 in only one allele in Japanese patients with LGMD2B.
In MM, patients with the c.2997G>T (p.Trp999Cys) mutation had a significantly late onset.13 Tagawa et al6 reported that age of onset in patients homozygous for the c.2997G>T (p.Trp999Cys) mutation was later than the third decade of life. In this study, especially in patients homozygous for the c.2997G>T (p.Trp999Cys) mutation, onset was significantly late. Only 10% of patients homozygous for the c.2997G>T (p.Trp999Cys) mutation developed muscle symptoms by 30 years. In contrast, 90% of patients without this mutation developed muscle symptoms by the same age. However, it is difficult to explain why the c.2997G>T (p.Trp999Cys) mutation is related to late onset forms. In an immunohistochemical analysis by Tagawa et al,6 although one patient with the homozygous c.2997G>T mutation (p.Trp999Cys) showed an ‘abnormal’ pattern, cytoplasmic accumulation of immunopositive material with deficiency of membrane staining or positive/negative mosaic membrane staining, three patients with the same mutation showed a negative pattern. Guglieri et al21 reported that patients carrying two truncating mutations showed the first muscular symptoms earlier in life than subjects harbouring double missense substitutions. Indeed, the c.2997G>T mutation is theoretically deduced to be a missense mutation (p.Trp999Cys). Meanwhile, in this study, there were only two patients carrying missense mutations other than the c.2997G>T (p.Trp999Cys) mutation. Thus it is difficult to discuss statistically whether an association between the c.2997G>T (p.Trp999Cys) mutation and late onset forms is due to a missense mutation or other inherent characteristics. Nonetheless, it is important that the late onset phenotype is found with prevalent mutations.
Although patients homozygous for the c.2997G>T (p.Trp999Cys) mutation had late onset, no difference in progression was observed in those harbouring the c.2997G>T (p.Trp999Cys) mutation, except for difficulty in standing on tiptoe. This suggests that the c.2997G>T (p.Trp999Cys) mutation is related to late onset but not to the slow progression of the disease. Furthermore, the c.2997G>T (p.Trp999Cys) mutation may be relevant to the proximal dominant impairment in dysferlinopathy, diagnosed as the limb girdle type. Interestingly, there was only one patient homozygous for the c.2997G>T (p.Trp999Cys) mutation in 27 families with MM.13 Moreover, patients homozygous for the c.2997G>T (p.Trp999Cys) mutation had a high probability that the muscle weakness started in the upper limbs. In addition, serum CK levels in patients homozygous for the c.2997G>T (p.Trp999Cys) mutation were lower than those in the other two groups.
We investigated the clinical features of 40 patients in 36 families with LGMD2B in whom dysferlin mutations were confirmed. Disease duration was long (26.6±9.9 years) in this study. The clinical features of LGMD2B in this study were as follows: (1) onset in the late teens or early adulthood except in patients homozygous for the c.2997G>T (p.Trp999Cys) mutation; (2) lower limb weakness at onset in most patients; (3) distal change of lower limbs on muscle CT in the early stage; (4) impairment of lumbar erector spinal muscles on muscle CT in the early stage; (5) predominant involvement of the proximal upper limbs; (6) preservation of function of the hands in late stage; (7) preservation of strength in the neck muscles in late stage; (8) lack of facial weakness or dysphagia; (9) avoidance of scoliosis; (10) hyper-Ckaemia; (11) preservation of cardiac function; and (12) tendency for respiratory function to decline with disease duration and occasional necessity for ventilatory assistance.
Age at onset was similar to that reported for various types of mutations.18 ,19 ,21 However, patients with disease onset at 73 years24 ,29 or congenital onset30 have been reported. Clinicians may not have taken into account analyses of the dysferlin gene for such patients.
Nishida et al proposed the name ‘distal limb girdle type muscular dystrophy’ for patients with MM that develop proximal muscle involvement relatively early.31 ,32 Nguyen et al17 classified patients for whom it was not possible to distinguish between a distal phenotype of MM and a limb girdle phenotype, even when examined at onset, as a distinct ‘proximodistal’ phenotype group. Although patients with the typical features of MM13 ,33 or asymptomatic hyper-CKaemia were excluded from the present study, variable patterns of weakness in the lower limbs were observed, as in previous studies.17 ,19 ,20 ,22 ,24 However, the predominant muscle weakness was in proximal sites of the upper limbs in the present study.
The results of muscle CT scan in this study were similar to those of detailed imaging studies in LGMD2B at a relatively early stage.23 ,34 ,35 In this study, we had only one case in whom a low density change in the dorsal muscle in the neck region occurred in the early stage. In contrast, two patients showed fatty muscle degeneration of the cervical elector spinae muscles after 9 or 10 years of disease duration, in a study performed using 3.0 T MRI.35 Another exceptional finding was a case in which the quadriceps femoris muscles were more severely damaged than the hamstrings, which was reported in two of five patients.34
In the present study, only two patients showed levels of EF that were <50% and the patient who showed the lowest EF (32.7%) had no symptoms or signs of cardiomyopathy.36 Guglieri et al21 reported cardiac rhythm changes in three patients and left ventricular hypertrophy in four patients from a total of 22 LGMD2B patients. Wenzel et al37 reported ECG abnormalities (repolarisation abnormalities or left ventricular hypertrophy) in four patients and pathological echocardiographic parameters in five of seven LGMD2B patients. Furthermore, two patients had symptoms and signs of dilated cardiomyopathy. Choi et al38 reported left ventricular hypertrophy on ECG in two patients, no cardiac signs in one patient and mildly decreased EF (45%) in one patient among five MM patients. Therefore, we think that in most patients with LGMD2B, in spite of the existence of laboratorial abnormalities, cardiac function was clinically preserved.
Mahjneh et al16 reported that patients with LGMD2B with disease durations of up to 7 years might show slight restrictive lung disease. Cagliani et al39 reported that respiratory tests showed mild obstructive signs at the small airways in a 20-year-old man with LGMD2B. Illa et al12 reported that pulmonary function tests revealed a mild reduction in VC in two patients with DACM. In this study, many patients showed pathological levels of respiratory parameters and levels of %VC decreased with disease duration. Furthermore, some patients needed NIPPV and one patient died of respiratory failure. The change in muscles related to respiratory function worsens with disease duration. Therefore, it is important to pay attention to respiratory function in patients with dysferlinopathy.
Acknowledgments
The authors thank the patients for participating in the study, Dr Mitsuru Kawai, National Hospital Organisation East Saitama National Hospital, for providing advice and Mr Brent Bell for reading the manuscript.
References
Footnotes
-
Contributors TT designed the study, conducted the mutational analysis, analysed the data, and drafted and revised the paper. MA supervised the study and revised the paper. NSu analysed the data, and drafted and revised the draft paper. MT analysed the data and revised the draft paper. CY, HSa, MH, HSu, MI, EA-K and NSh performed the mutational analysis and revised the draft paper. TI, SKur, TW, GS, NF, TS, TM, IF, EM, TK, MM, MYa, TH, JS, ST, SKuz and HT analysed the data and revised the draft paper. MYo, HK, HO and YI supervised the study and revised the draft paper.
-
Funding This work was supported by Intramural Research Grants (20B-12, 20B-13, 23-4 and 23-5) for Neurological and Psychiatric Disorders of NCNP.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval The study was approved by the National Hospital Organisation Nishitaga National Hospital and Tohoku University School of Medicine.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/