Article Text

Abstract
Objective To report the clinical and investigative features of children with a clinical diagnosis of probable autoimmune encephalopathy, both with and without antibodies to central nervous system antigens.
Method Patients with encephalopathy plus one or more of neuropsychiatric symptoms, seizures, movement disorder or cognitive dysfunction, were identified from 111 paediatric serum samples referred from five tertiary paediatric neurology centres to Oxford for antibody testing in 2007–2010. A blinded clinical review panel identified 48 patients with a diagnosis of probable autoimmune encephalitis whose features are described. All samples were tested/retested for antibodies to N-methyl-D-aspartate receptor (NMDAR), VGKC-complex, LGI1, CASPR2 and contactin-2, GlyR, D1R, D2R, AMPAR, GABA(B)R and glutamic acid decarboxylase.
Results Seizures (83%), behavioural change (63%), confusion (50%), movement disorder (38%) and hallucinations (25%) were common. 52% required intensive care support for seizure control or profound encephalopathy. An acute infective organism (15%) or abnormal cerebrospinal fluid (32%), EEG (70%) or MRI (37%) abnormalities were found. One 14-year-old girl had an ovarian teratoma. Serum antibodies were detected in 21/48 (44%) patients: NMDAR 13/48 (27%), VGKC-complex 7/48(15%) and GlyR 1/48(2%). Antibody negative patients shared similar clinical features to those who had specific antibodies detected. 18/34 patients (52%) who received immunotherapy made a complete recovery compared to 4/14 (28%) who were not treated; reductions in modified Rankin Scale for children scores were more common following immunotherapies. Antibody status did not appear to influence the treatment effect.
Conclusions Our study outlines the common clinical and paraclinical features of children and adolescents with probable autoimmune encephalopathies. These patients, irrespective of positivity for the known antibody targets, appeared to benefit from immunotherapies and further antibody targets may be defined in the future.
- Limbic System
- Movement Disorders
- Amnesia
- Paediatric Neurology
- Epilepsy
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Encephalopathy in children and adolescents is associated with a high rate of morbidity and mortality and poses difficult diagnostic and therapeutic challenges.1 ,2 The differential diagnoses are diverse, including infectious, para-infectious, metabolic, genetic, traumatic, malignant and toxic disorders.1 ,3 The clinical features of these disorders overlap and, in many cases, the cause will not be apparent from the history and examination at presentation. Clinical observation, investigation and treatment often need to be carried out simultaneously.1
Antibody-mediated encephalopathies, in which patients present with neurological syndromes associated with serum and/or cerebrospinal fluid (CSF) antibodies directed against ion channels, receptors and associated proteins, are now well recognised in adults.4–6 Although the antigenic targets are generally expressed throughout the nervous system, the clinical picture can localise to specific areas of the central nervous system (CNS). Many patients present with amnesia, confusion, seizures and psychiatric features,4 and some then develop a more generalised encephalopathy with a movement disorder.5 In some patients the encephalopathy can be part of a paraneoplastic neurological disorder; the presence of a tumour depends not only on the type of autoimmune disorder, but also on the age and gender. Nevertheless, the majority of patients do not have detectable tumours.4
Autoimmune encephalopathies are increasingly being diagnosed in children with antibodies to N-methyl-D-aspartate receptor (NMDAR),7–10 and in some with antibodies to voltage-gated potassium channel (VGKC)-complex proteins11–17 or other CNS antigens such as glutamic acid decarboxylase (GAD).14 In a large US cohort,7 40% of NMDAR antibody encephalitis presented in childhood or adolescence, and in a smaller study from the UK, 23% of patients were under the age of 18.8 Additionally, in a multicentre UK encephalitis study,18 16/75 (21%) of patients without a detected infection were found retrospectively to have a specific antibody, a frequency that was higher than any individual virus identified. A similar observation regarding NMDAR antibodies was made in the Californian encephalitis project.19
The clinical phenotypes associated with these conditions are increasingly recognised but some patients are negative for the available antibody tests. In order to assess the clinical and investigative features, immunotherapy responses and outcomes, in patients with specific antibodies, and to compare them with those in whom no antibodies were detected, we studied patients whose sera were referred from five paediatric neurology centres, and selected for study those in whom the final diagnosis was of a probable autoimmune encephalopathy.
Methods
Patient selection and review of immunotherapy response
The children, all under 18 years, were seen at the Evelina Children's Hospital, London (n=36), Great Ormond Street Children's Hospital, London (n=30), Birmingham Children's Hospital, Birmingham (n=28), St George's Hospital, London (n=10) and King's College Hospital, London (n=7) between 2007 and 2010. Serum samples from these 111 patients were sent to the Oxford laboratory for testing for CNS autoantibodies. These children underwent extensive diagnostic studies as indicated by the clinical history to exclude infective, genetic and metabolic disorders. Clinical data, comprising demographic information, clinical features at presentation, discharge and follow-up, and results of laboratory, electrophysiological and neuroimaging testing, were compiled (YH and SW), and presented anonymously, and without revealing available antibody positivity, to a multicentre review panel of four paediatric neurologists (AC, CH, EW, ML). Patients were considered to have a probable immune-mediated encephalopathy if they presented with encephalopathy (depressed or altered level of consciousness, lasting more than 24 h, lethargy, or change in personality or behaviour) as previously defined,18 and at least one of the following features: neuropsychiatric symptoms, seizures, movement disorder, or cognitive dysfunction. Patients were excluded if an alternative diagnosis was made but included even if the diagnosis of probable autoimmune encephalopathy was made retrospectively. In addition, we chose to exclude patients with demyelinating disease (who are being studied separately) and two with Rasmussen encephalitis, as well as those whose encephalopathy was given an alternative diagnosis (see figure 1 for details). An immunotherapy response was defined as ‘definite’ if the response could not be attributed to another factor or when patients were treatment-dependent and relapsed on discontinuation. A ‘probable’ response was when a confounder such as antiepileptic drugs or possible spontaneous improvement could not be excluded. The outcomes, as measured by the range of difficulties the patients were experiencing (cognitive, behaviour, seizure and motor), were retrieved from the patient's medical records or, if unavailable, were obtained directly from the patient's primary paediatric neurologist. Full recovery was defined by the absence of medical, educational and social concerns reported from parents, school and the patient's primary clinician at follow-up (minimum follow-up time of 1 year). We also applied the modified Rankin Scale (mRS) for children, as previously used by Bigi et al,20 to assess the degree of disability both during the acute admission and at follow-up. Ethical approval for this study was from the Oxfordshire Regional Ethical Committee A (07/Q1604/28).
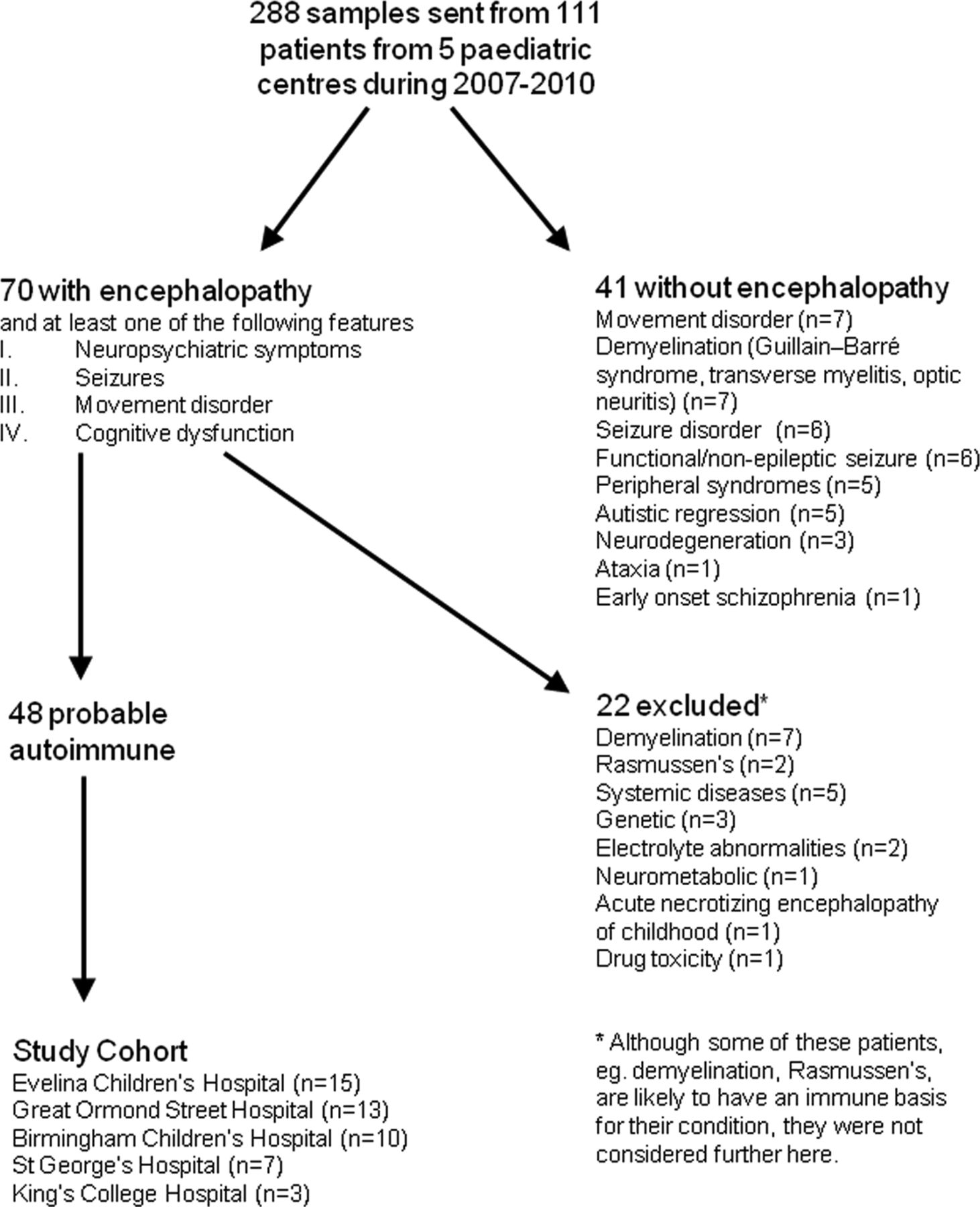
Study flow chart showing expert review and record linkage outcome. Four patients who were excluded from the study because they were not encephalopathic were positive for autoantibodies in the serum; two had VGKC-complex antibodies (one Guillain–Barré syndrome and one myositis) and two had N-methyl-D-aspartate receptor antibodies (one movement disorder, one optic neuritis). Patients with demyelinating conditions, Rasmussen encephalitis, or with neurological symptoms secondary to systemic diseases could have an autoimmune mechanism for their disorder but were excluded from this study.
Autoantibody testing
All samples were tested originally for VGKC-complex or NMDAR antibodies as requested. Retrieved sera, and the four CSFs sent, were retested for all available antibodies. Serum (at 1:20 dilution) antibodies to NMDAR, leucine-rich glioma inactivated 1 (LGI1), contactin-associated protein 2 (CASPR2; serum at 1:100), contactin-2, glycine receptor (GlyR), and the dopamine receptors 1 and 2 (D1R and D2R) were tested using cell-based assays in routine clinical use. D1R and D2R full-length human dopamine receptors were individually subcloned from image clones into the mammalian expression vector pcDNA-DEST40. Monoclonal antibodies (anti-D1 dopamine receptor produced in rat, Sigma-Aldrich D2944; anti-DRD2 antibody produced in mouse, WH0001813M1 SIGMA), confirmed good surface expression of the antigens. For all the cell-based assays, the binding of serum IgG to the surface of human embryonic kidney cells, transfected with cDNA encoding the auto-antigen, is visualised using a fluorescence-labelled secondary antibody. The binding is scored visually on a range from 0 (negative) to 4 (very strong) as in previous studies.8 ,21 ,22 AMPA receptor (AMPAR) and GABA(B)R antibodies were tested using a commercial assay (Euroimmun, Luebeck, Germany) and negativity confirmed with in-house tests. VGKC-complex antibodies were measured by immunoprecipitation of rabbit brain VGKC-complexes labelled with radioactive dendrotoxin (normal <100 pM, low positive 100–150 pM, positive 150–400 pM and high positive >400 pM).23 GAD antibodies were measured by immunoprecipitation of 125I-recombinant GAD.24
Data analysis
Descriptive statistics were used to summarise the key components of patient data. Fisher's exact test (two-tailed) was used for comparison of clinical details and investigatory findings in the different groups. The χ2 test was used to assess the relationship between clinical improvement and use of immunotherapies. Data were analysed using GraphPad Prism 5.
Results
Clinical features of the patients and relationship to autoantibodies
Following the multicentre expert review panel, 48/111 patients were considered to have a diagnosis of probable autoimmune encephalopathy (see figure 1). Of the 48 patients, 26 were female (mean 8.7 years, range 1.83–17) and 22 were male (mean 9 years, range 0.16–16). The ages of onset and patients’ demographics are shown in figure 2. Non-Caucasians were common, representing 24/48 (50%) of the cohort.
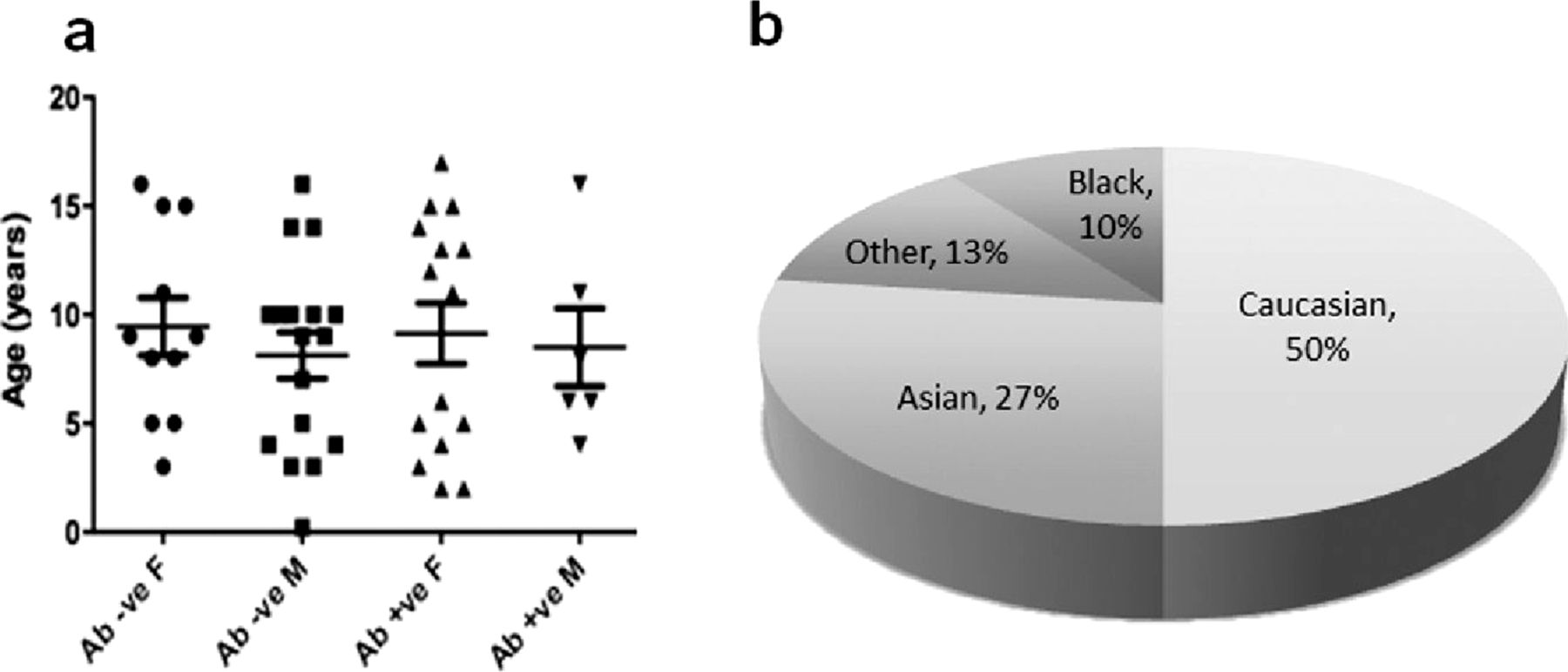
Demographics of patients with autoimmune encephalopathy. (A) Age at disease onset of the 48 paediatric patients with autoimmune encephalopathy divided between antibody positive and antibody negative, male and female patients. (B) Ethnicity of the 48 paediatric patients with autoimmune encephalopathy; 24 patients (50%) were non-Caucasian: Asian (13), Black (5) and Other (6).
Prodromal symptoms, with either fever and/or associated infectious episodes, were noted in 27 patients (56%). Three patients had a recent vaccination (Pandemrix for H1N1, HPV and Men C booster; 1 month, 4 weeks and 1 week prior to onset, respectively). The most common presenting features included seizures (83%; generalised seizures 71%, focal seizures 56%), behavioural changes (63%) and confusion (50%). Twenty-seven patients had neuropsychiatric symptoms (56%; agitation 38%, hallucinations 25%) and 18 developed a movement disorder (38%; choreoathetosis 29%, myoclonus 2%, startle 4%, tremor 4%). Twenty-five patients (52%) required intensive care admission for seizure control or reduction in conscious level.
Forty-five patients had NMDAR antibodies requested and 32 patients had VGKC-complex requested initially; positive results were confirmed on retesting. Overall, 21 patients were antibody positive; 13 patients (27%, mean age 8.9, range 1.83–17) were positive for NMDAR antibodies, with cell-based assay scores of 2.5–4 (controls <1); one of the two CSFs was also positive. Sera from seven different patients (17%, mean age 9, range 4–15) immunoprecipitated VGKC-complexes, with results ranging from 105 to 1360 pM; the only CSF available for testing was negative for these antibodies. Two of the VGKC-complex antibody patients were also positive for GAD antibodies, one of them with type 1 diabetes mellitus, but none were positive for LGI1, CASPR2 or contactin-2 antibodies. A 5-year-old girl who presented with explosive-onset epileptic encephalopathy (without rigidity and myoclonus) was found to have GlyR antibodies in her serum (score 2.5) and CSF (score 1). None of the patients had antibodies to AMPAR, GABA(B)R (confirmed subsequently with in-house assays), D1R or D2R. Table 1 demonstrates no apparent differences in clinical features between the antibody positive and negative groups. However, when the clinical features of patients with NMDAR antibodies and VGKC-complex antibodies were compared, there was a trend towards more frequent movement disorders and hallucinations with the NMDAR antibodies (table 2).
Comparison of the clinical features and association with infections in paediatric patients with antibody positive and antibody negative encephalopathy
Comparison of clinical and investigatory findings of patients with VGKC-complex and NMDAR encephalitis
CSF analysis, neuroimaging and electrophysiological studies
CSF analysis revealed lymphocytosis in 8/47 (17%) patients and oligoclonal bands in 6/47 (13%, table 3). Interestingly, three patients had organisms identified in the CSF (two herpes simplex virus (HSV), one adenovirus); the two positive for HSV became progressively worse despite adequate antiviral therapy and were found to have NMDAR antibodies when reinvestigated.
Comparison of the paraclinical features in paediatric patients with antibody positive and negative encephalopathy
Brain imaging was normal in 35/48 (73%) at presentation and the remaining patients had increased T2 changes in the cortex (n=6), hippocampus (n=3), white matter (n=3) or basal ganglia (n=1). These changes had normalised by 3 months but five patients developed atrophy on a repeat scan (table 3). Reversible MRI changes, without atrophy, were common in the VGKC-complex antibody patients. By contrast, the majority of the NMDAR antibody group had a normal MRI at the onset of the disease, but 4/13 developed atrophy by 6 months (table 2). One 14-year-old girl, who was positive for NMDAR antibodies, had an ovarian teratoma identified by ultrasound.
EEG, at presentation, demonstrated slow background consistent with an encephalopathy in 33/47 (70%) patients. In 10 others, the EEG demonstrated epileptiform discharges early in the disease course; seven of these progressed to an encephalopathic pattern. In two patients there was slowing of alpha and increased beta activity, probably due to benzodiazepine use.
Response to immunotherapy in patients with autoimmune encephalitis
Immunotherapy was given in 34/48 (73%). First-line immunotherapies (corticosteroids, intravenous immunoglobulins (IVIG) and plasma exchange (PLEX)) were given in 29 patients, and five required regular immunosuppressive treatment (azathioprine, cyclophosphamide, rituximab, mycophenolate). Two patients received immunotherapy late in the disease (more than 6 months from the onset of symptoms) and did not respond to treatment.
Of the patients who did not receive immunotherapy, only 4/14 (29%) recovered completely compared to 18/34 (53%) who received immunotherapy, although this difference did not reach statistical significance. Both HSV/NMDAR antibody positive children showed a definite response to intensive immunotherapy with steroids, IVIG and PLEX. Overall 32/34 (94%) showed some response to immunotherapy; 23/34 (68%) ‘probably’ responded (7 NMDAR, 3 VGKC-complex, 13 seronegative), with only 9/34 (28%) showing a ‘definite’ response (5 NMDAR, 1 VGKC-complex, 3 seronegative) (see earlier definitions), but there was no apparent difference in treatment responses between the antibody positive and the antibody negative groups (table 4).
Comparison of immunotherapy response and outcome in paediatric patients with antibody positive and antibody negative encephalopathy
Medium term outcome of patients
At the latest follow-up (12–60 months from onset, mean 24 months), 20/48 (42%) patients have made a complete recovery, 24 (50%) still have cognitive and behavioural problems, and 16 (33%) have ongoing seizures with one child still severely disabled (figure 3). The outcomes in the antibody positive group have been slightly worse, with only 6/21 (29%) compared to 14/27(52%) in the antibody negative group making a complete recovery. Nevertheless, the mRS for children scores demonstrated a significant change between nadir and final follow-up in the group as a whole, with trends in both the antibody positive and negative groups (table 4 and figure 4).
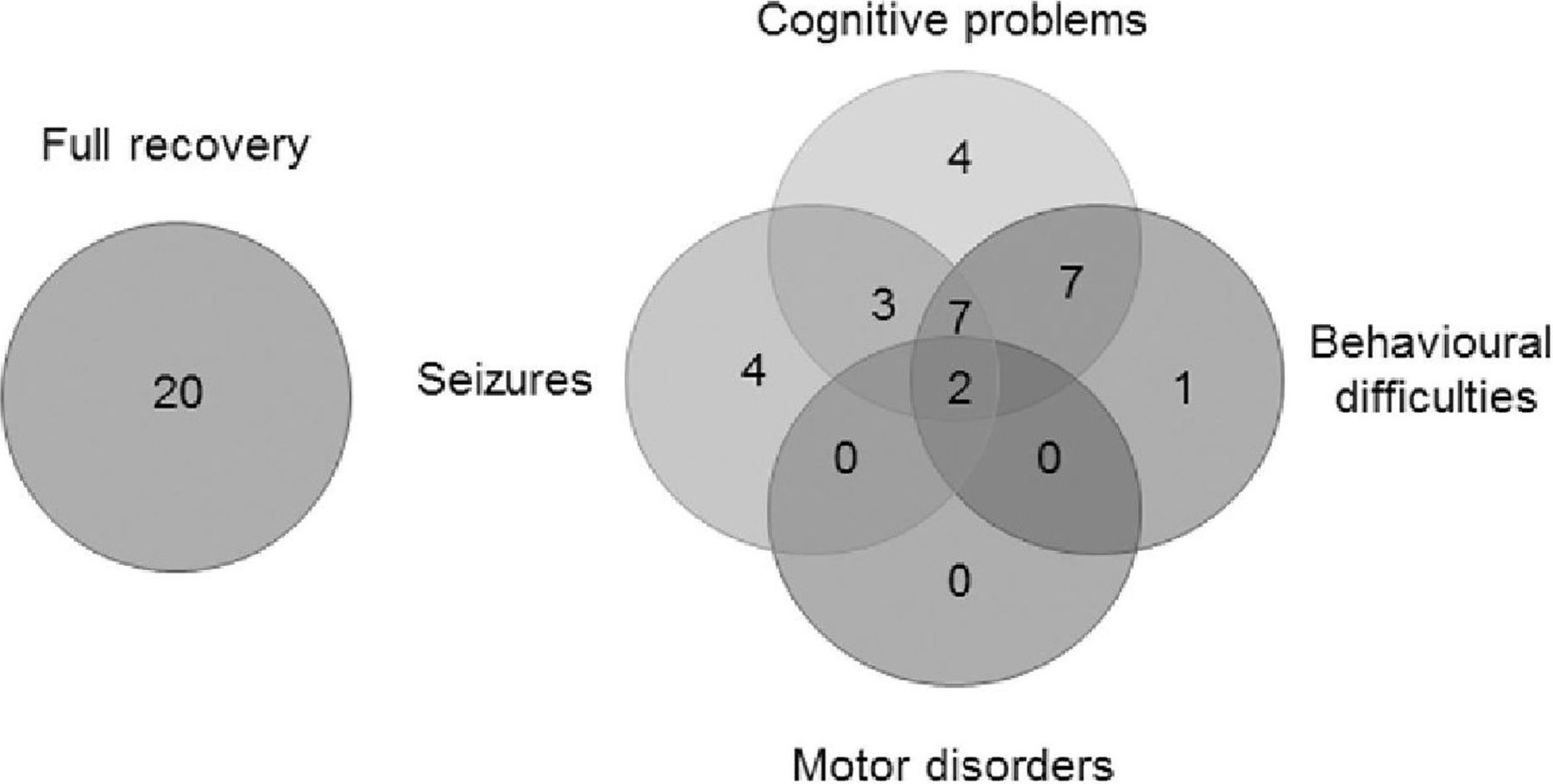
Outcome of all patients with autoimmune encephalopathy. The problems encountered in children at follow-up of 1–5 years (mean=24 months). Twenty patients recovered completely. Cognitive problems were still present in 23, seizures in 16, behavioural problems in 17 and two patients had additional motor problems.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Modified Rankin Scale (mRS) for children score reduction in total cohort and in the antibody positive and negative groups, stratified according to immunotherapies. A significant improvement of the mRS score between nadir and final follow-up, as measured by the χ2 test, was seen in the patients who received immunotherapies (p=0.04) (A), with a similar trend in the antibody positive (B) and antibody negative (C) groups.
Discussion
Autoimmune forms of encephalopathies are now well recognised in children as well as adults, but there are few comparative studies of their clinical and investigative features or outcomes, and it is not clear what proportions of patients have antibodies to the CNS targets that have only been identified in the last few years. In a group of patients with probable diagnosis of autoimmune encephalopathy, defined by their clinical features and exclusion of other known causes, we found that 21 (46%) had antibodies to either the NMDAR (27%, one with an ovarian teratoma) or the VGKC-complex (17%), and one (2%) to GlyR antibodies. No significant clinical differences were found between the antibody positive and negative groups. Moreover, both antibody positive and negative patients responded to immunotherapies with improvements in the mRS scores and complete recovery in 20/48 (42%) overall. Thus it appears that the lack of identification of a known antibody by current available methods should not exclude the diagnosis of autoimmune encephalopathy, and immunotherapy could be considered. Future investigations of the antibody negative group may lead to the definition of new antigenic targets.
The clinical presentation of patients with NMDAR antibodies (table 2) is consistent with those previously described in the adult5 ,8 ,25 and paediatric literature7 ,9 ,10; the existence of only one patient with a teratoma likely reflects the young age of the children studied.5 Children with VGKC-complex antibodies presented either with limbic encephalitis and prolonged seizures, or with a less specific encephalopathy syndrome with varying degrees of seizures and psychiatric features, as previously reported in smaller studies.11 ,13 ,14 Only one child had an unexpected positive GlyR antibody which is more commonly associated with a progressive or relapsing remitting form of stiff person syndrome, often with additional brainstem and autonomic features and mainly, but not exclusively, in adults (Vincent A, unpublished observations).22 ,26 ,27
All recruiting centres undertook investigative studies to exclude infective aetiologies, often with support from paediatric infectious disease teams. Nevertheless, within the antibody positive group, 5/21 (24%) patients were positive for an acute infective organism (HSV (2), mycoplasma (2), Epstein–Barr virus (1)); three patients had a raised anti-streptococcal antibody titre, and yet phenotypically they resembled NMDAR antibody encephalitis. Indeed mycoplasma IgM positive serology has previously been reported in 50% of patients with NMDAR without a neoplasm.28 Moreover, the two NMDAR antibody positive patients who were also positive for HSV PCR in the CSF, continued to regress despite acyclovir treatment but improved following immunotherapy. It is possible that some patients have existing antibodies that gain access to the brain when there is a viral infection, or alternatively a para-infectious antibody-mediated process may begin during an acute infective process. Consistent with this, NMDAR antibodies have recently been reported in 30% of patients with HSV encephalitis, possibly as a secondary epiphenomenon.29
Generalised MRI atrophy was reported in 5/48 (10%) of our patients. The small sample size did not allow us to correlate the MRI findings with symptom severity or outcome. Postmortem reports of some NMDAR encephalitis patients showed pronounced brain atrophy, although reversible atrophy was observed in others who survived.30 By contrast, a recent study of immunopathology of antibody-associated encephalopathies including NMDAR encephalitis showed lack of neuro-axonal injury with minimal inflammation in the cortex and no atrophy on imaging.31 The cause of the atrophy in our patients, in contrast to the minimal changes on imaging, is unclear and could be a result of inflammation, recurrent seizures or medication (corticosteroids). Initial EEGs were encephalopathic or showed a slow background in the majority of the patients, in keeping with previous reports.7 ,9 ,12 Generalised seizures were most frequently reported, but it is possible that focal seizures were masked by the encephalopathic changes.
Immunosuppressive strategies with corticosteroids, IVIG and PLEX have been used extensively in adults and are beginning to be used more commonly in children.32 Responses to immunotherapy, particularly for NMDAR antibody encephalitis, can be slow (over months) and variable, often requiring a multidisciplinary team for detailed management. Furthermore, the latency to treatment is also very variable (months to years) as in previously-reported childhood series,7 ,9 ,10 ,12–14 making interpretation and direct comparison of treatment efficacy difficult. Although complete recovery was seen in 42% of our patients, it is important to note that learning, memory and behavioural problems were often not identified until the child was integrated back into the educational system. NMDAR antibody associated encephalitis can relapse in approximately 30% of adults5 ,8 and 25% of children,7 ,10 and the need for longer-term immunosuppression, currently maintained in only five of our patients, will have to be considered when more data are available.
In general in our cohort, worse outcomes were seen in patients who did not receive any immunotherapy, and delay to therapy was likely to have influenced outcome, as seen in the two patients who received immunotherapy late and did not respond. Nevertheless, four patients who did not receive immunotherapy made a complete recovery. Spontaneous recoveries in immune-mediated encephalopathies have been described,12 ,17 ,33 and suggest that some of the clinical syndromes may be monophasic and resolve without treatment. Treatment decisions must balance the potential benefits of early and sustained immunotherapy in prevention of progression of disease and in facilitating recovery.
The presence of an antibody negative group raises questions about our selection process and the diagnosis in such patients. First, the lag phase from clinical presentation to antibody testing, or sampling after immunotherapy, may explain negative tests in some of the patients. Although rare patients have antibodies in CSF but not in serum,5 in this laboratory's experience, CSF, diluted 1:1, is never positive when the serum, diluted 1:20, is negative; indeed in the four cases with CSF available, the serum levels were clearly higher than the CSF levels, two of which were negative; thus lack of CSF testing is unlikely to be a factor. Second, two of the antibody negative patients had their serum sample taken after receiving IVIG which might have influenced their results, but since 16/18 antibody positive patients remained positive in the serum at 1 year from symptoms onset (11/12 NMDAR, 4/5 VGKC-complex and 1/1 GlyR), despite immunotherapies, it is more likely that the antibody negative patients have an unidentified CNS auto-antigen or possibly some other form of immune-mediated disease. Given that some of the antibody negative patients had movement disorders, we looked for antibodies to the dopamine receptors D1R and D2R. Antibodies to D2R have recently been reported in a paediatric cohort with basal ganglia encephalitis.34 Those patients were phenotypically similar to a few of the antibody negative patients in our cohort, but we were not able to find any dopamine receptor antibodies in their sera.
In this systematic study of patients referred to tertiary centres, only 44% of patients with an eventual diagnosis of probable autoimmune encephalopathy were positive for known autoantibodies. These patients were often apyrexial at presentation, had low CSF cell counts and did not have MRI abnormalities that were characteristic of identifiable viral encephalitis, features that have helped to distinguish viral from immune aetiologies in previous studies.18 ,28 The similarities between patients with and without identified antibodies indicate a need to search for new antigenic targets that could be helpful in future diagnosis; in the meantime diagnostic and management strategies that are often based on antibody evaluation, and directed by one of a specific antibody, may need to be re-evaluated.
Acknowledgments
We thank Dr Bethan Lang, Dr Leslie Jacobson and Ms Anjan Nibber for confirming antibody results for AMPAR, VGKC-complex, GAD and NMDAR antibody results.
References
Footnotes
-
Contributors The study was designed by YH, ML and AV, with suggestions from SW. PW and AV were responsible for supervising the antibody assays. CH, AC, EW and ML were respective principle investigators from their institutions. ML and AV supervised the study as a whole. All authors contributed to the evaluation and recording of clinical data, and to the writing of the manuscript.
-
Funding This work was supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre based at Oxford University Hospitals NHS Trust and the University of Oxford (YH, PW and AV) and the Wellcome Trust (SW).
-
Competing interests AV and the University of Oxford hold patents and receives royalties and payments for antibody assays. No other competing interests identified.
-
Ethics approval Oxfordshire Regional Ethical Committee A.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Linked Articles
- Editorial commentaries