Article Text

Abstract
Background Autosomal dominant (AD) central core disease (CCD) is a congenital myopathy characterised by the presence of cores in the muscle fibres which correspond to broad areas of myofibrils disorganisation, Z-line streaming and lack of mitochondria. Heterozygous mutations in the RYR1 gene were observed in the large majority of AD-CCD families; however, this gene was excluded in some of AD-CCD families.
Objective To enlarge the genetic spectrum of AD-CCD demonstrating mutations in an additional gene.
Patients and methods Four affected AD family members over three generations, three of whom were alive and participate in the study: the mother and two of three siblings. The symptoms began during the early childhood with mild delayed motor development. Later they developed mainly tibialis anterior weakness, hypertrophy of calves and significant weakness (amyotrophic) of quadriceps. No cardiac or ocular involvement was noted.
Results The muscle biopsies sections showed a particular pattern: eccentric cores in type 1 fibres, associated with type 1 predominance. Most cores have abrupt borders. Electron microscopy confirmed the presence of both unstructured and structured cores. Exome sequencing analysis identified a novel heterozygous missense mutation p.Leu1723Pro in MYH7 segregating with the disease and affecting a conserved residue in the myosin tail domain.
Conclusions We describe MYH7 as an additional causative gene for AD-CCD. These findings have important implications for diagnosis and future investigations of AD-congenital myopathies with cores, without cardiomyopathy, but presenting a particular involvement of distal and quadriceps muscles.
- MYOPATHY
- MUSCLE DISEASE
- NEUROPATHOLOGY, MUSCLE
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Autosomal dominant (AD) central core disease (CCD) is an inherited disorder characterised by the presence of cores constituted by abnormally compacted myofibrils, Z-band streaming and absence of mitochondria. Classically, the cores are well-limited rounded areas devoid of any oxidative staining and they extend almost along the entire length of the fibres. Genetic studies of most of the AD-CCD families demonstrated the presence of heterozygous mutations in the RYR1 (ryanodine receptor type 1) gene predominantly located in the calcium channel at the C-ter domain. However, RYR1 was excluded in some AD-CCD families, suggesting a genetic heterogeneity.1 Moreover, different patterns of cores with abrupt borders could be observed in transverse muscle sections from patients affected by AD-core disease such as ‘central cores’, ‘eccentric cores’, and single or multiple ‘peripheral cores’.1 ,2
Mutations of the MYH7 gene encoding the muscle heavy chain myosin (muscle slow/β cardiac MyHC) cause several and clinically diverse pathological conditions, mainly familial hypertrophic/dilated cardiomyopathy (MIM 613426, MIM 192600), as well as different skeletal muscle disorders as Laing early-onset distal myopathy (MIM 160500), myosin storage myopathy (MIM 608358), scapuloperoneal and limb girdle syndromes, and other disorders including cardiomyopathies with ‘core-like’ lesions.3 MYH7 mutations associated with cardiomyopathies are generally clustered in the myosin motor domain or calmodulin binding domain, while mutations associated with pure myopathies mainly reside in the myosin tail domain.3–6 Some previously reported patients with MYH7 mutations displayed both skeletal and cardiac muscles involvement.7–10
Here we report a family with AD-eccentric core disease (ECD) without evidence of cardiomyopathy presented with a main distal and quadriceps involvement with hypertrophy of calves caused by a novel heterozygous MYH7 mutation.
Clinical findings
For 28 years we followed a family of three living affected members: two siblings, a girl now 38 years old (P.III-2) and a boy now 36 years old (P.III-3), and their mother now 70 years old (P.II-2) (figure 1A). Clinical history started during childhood, with difficulties in sport activities and slight motor delay. All patients presented a very slowl progressive clinical course and a common phenotype consisting of tibialis anterior and peroneal (globally MRC grade 3/5), finger extensor and proximal lower limb weakness (quadriceps strength scoring 4/5). P.III-2 always presented with muscle pain after effort and global muscle fatigability. No myoglobinuria was reported. P.III-3 is the less affected and currently only presents with muscle fatigability. P.II-2 at 70 years old also presented with scapular winging, mild neck flexor weakness and sternocleidomastoid muscle atrophy. All patients presented pseudo-hypertrophic calves and developed Achilles tendon contractures. Facial and extraocular muscle were spared. No respiratory or cardiac (ECG and echocardiography) involvement was revealed. Serum creatine kinase (CK) levels were normal.
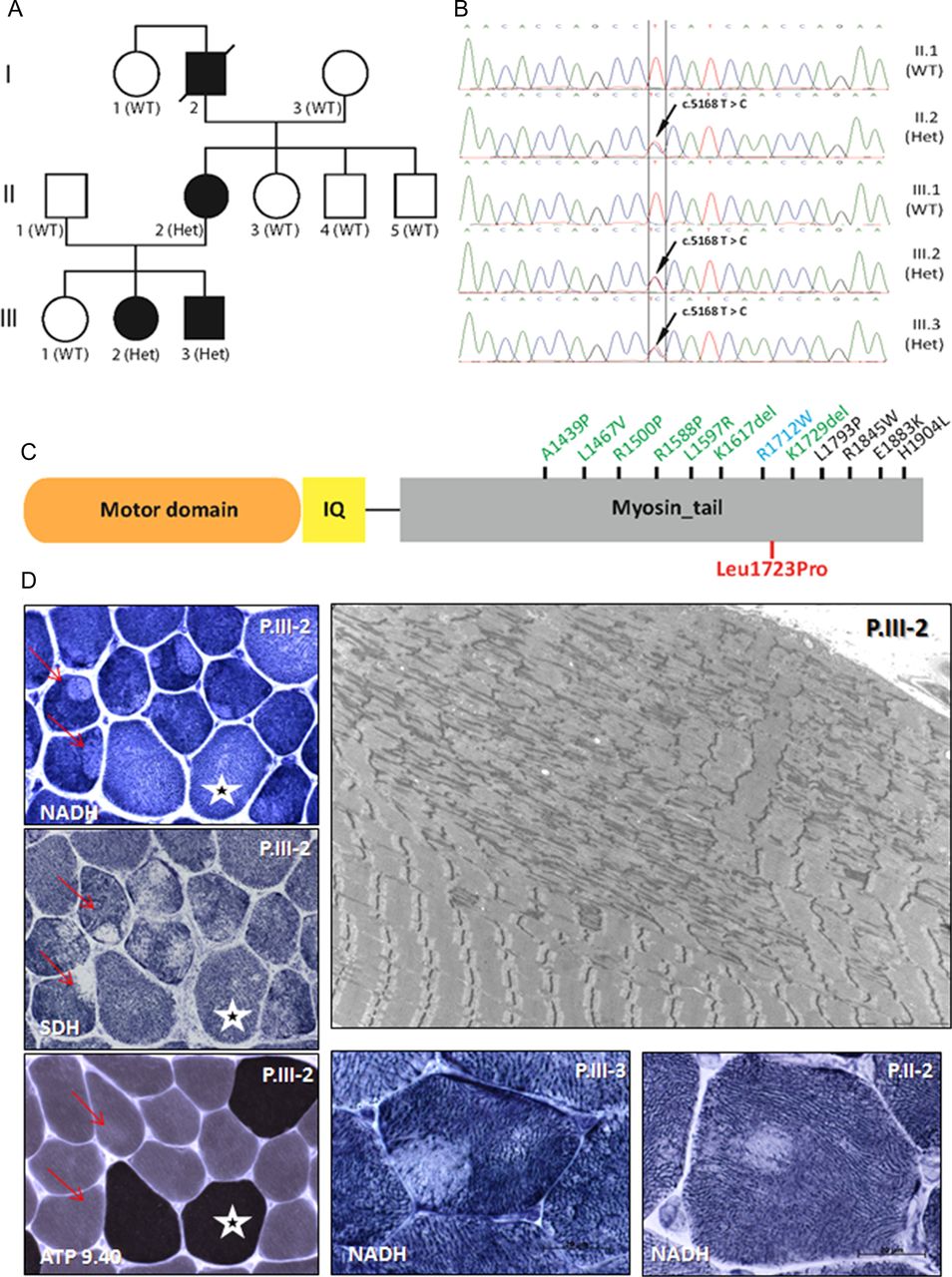
Characterisation of the autosomal dominant (AD)-eccentric core disease patients with MYH7 mutation. (A) Pedigree of the family and segregation of the MYH7 heterozygous mutation (Het) in the affected individuals. (B) Chromatopherograms from the Sanger sequencing showing the c.5168T>C mutation. (C) MYH7 protein domains (myosin motor domain or globular head; IQ, calmodulin binding domain; myosin tail or rod domain; not to scale) and reported mutations in the tail domain linked to cardiomyopathy (blue), myosin storage disease (black) or Laing myopathy or related myopathies (green). Mutations in the motor domain that are all linked to cardiomyopathy are not displayed. The p.L1723P AD-central core disease mutation described in this study is in red. (D) Muscle biopsies findings: The patient is indicated in the upper right corner and the histoenzymological techniques in the bottom left corner. Cryostat sections with oxidative enzyme reactions demonstrated the presence of eccentric cores, with well-delimited borders, in type I muscle fibres (red arrows). Type I fibres size are slightly smaller than type II (white stars). Electron micrograph from patient P.III-2 shows widespread subsarcolemmal unstructured cores with compaction of myofibrils, sarcomeric disorganisation, Z-band streaming and lack of mitochondria.
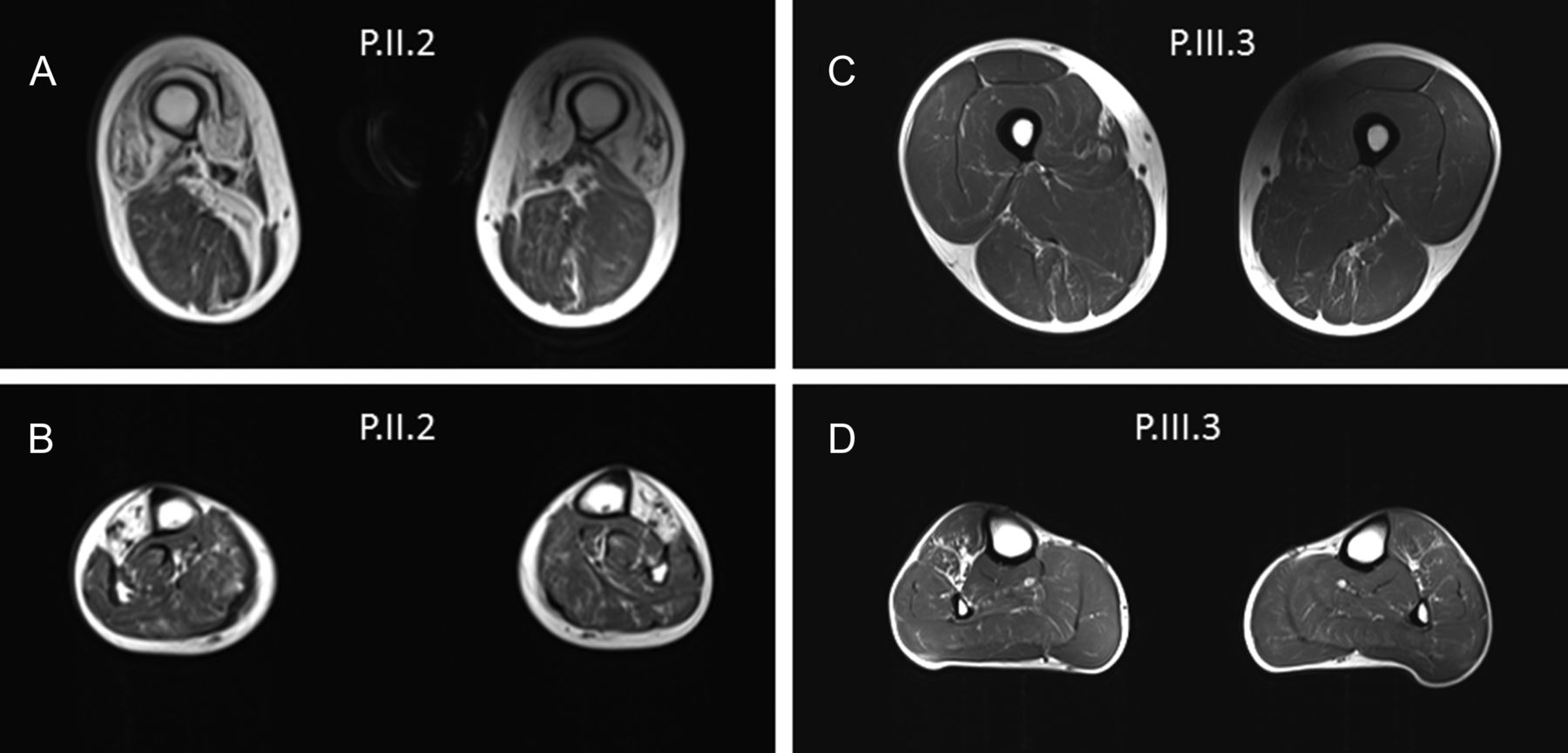
CT scan and MRI of lower limbs performed in these patients revealed that the most affected muscles were tibialis anterior (early), peroneal, quadriceps, glutaeus medius and lateral gastrocnemii. The MRI from P.II-2 performed at 70 years of age showed an almost complete involution of muscles of the anterior compartment of the legs (muscles tibialis anterior), a marked adipose involution of the anterior compartment of the thigh affecting almost all of the quadriceps on its entire length and a fatty replacement of internal compartment of the thighs. On the other hand, MRI from P.III-3 performed at 36 years of age showed only discrete fatty degeneration of the adductor longus and the anterior compartment of the legs with discrete right side predominance (figure 2).

{kind=link}
{kind=link}
Muscle MRI from P.II.2 performed at 70 years old showed a severe and selective involvement of the muscles of the anterior compartment of the thigh mainly in quadriceps (A) and anterior compartment of the legs predominantly in tibial anterior (B). Muscle MRI from P.III.3 performed at 36 years old showed a selective mild involvement of the anterior compartment of the legs mainly tibial anterior (D). Very mild abnormalities of the thigh were also noted (C).
Muscle morphology
Deltoid muscle (P.III-2 and P.III-3) and radialis muscle (P.II-2) biopsies were obtained at 12, 10 and 42 years of age, respectively. Type 1 fibre predominance (88%, 86% and 76%) and relative smallness (average 39 µm) was consistently observed. Oxidative enzyme reactions revealed the presence of ‘eccentric cores’ in type 1 fibres in the three biopsies. Most of these presented abrupt edges. There was no increased number of internalised nuclei. No necrotic/regenerative fibres or fibres IIC were observed. Ultrastructural examination confirmed the presence of both ‘structured’ and ‘unstructured’ cores, the latter corresponding to large areas of myofibrillar compaction, sarcomeric disorganisation and devoid of mitochondria (figure 1D). The cores extended along a great number of sarcomeres, almost along the full length of the fibres. Z-line streaming and focal sarcomere disruption were also noted in other areas; some of them were included in more large disorganised regions. In rare fibres, there was dilatation of the tubular reticulum system.
Molecular analysis
Linkage analysis excluded the RYR1 locus on chromosome 19. To identify the causative mutation for this AD-CCD family, we performed exome enrichment with the Agilent SureSelect Human All Exon 50 Mb kit followed by paired end sequencing from DNA of P.II-2 and P.III-3. We identified a novel variant common to both patients in the MYH7 gene: a heterozygous c.5168T>C transition in exon 36 leading to a change from an evolutionary conserved leucine to proline (p.Leu1723Pro) in the myosin tail domain (figure 1B,C). The schematic representation of the MYH7 protein shows that missense mutations in this region have previously been associated with Laing distal myopathy, cardiomyopathy or myosin storage disease. PCR and Sanger sequencing (primers 5′CTCCTGCCCCTAGGTTACTG3′ and 5′CACGGAGAGACACTGGTCTG3′) of 10 family members available confirmed the segregation of the heterozygous variant with the disease (figure 1A,B). The MYH7 p.L1723P mutation was found in the three tested affected individuals but in none of the healthy family members or in databases listing human genome variations as dbSNP, 1000 Genomes or exome variant server. The missense mutation is predicted to be damaging by sorting tolerant from intolerant (SIFT) and PolyPhen. Exome sequence analysis did not reveal any other potentially pathogenic variation in known muscle disorder genes.
Discussion
We report an AD-ECD family in which the gene RYR1 was excluded. We identified the MYH7 p.Leu1723Pro mutation following an integrated strategy combining exome sequencing with clinical and histopathological investigations.11 This missense mutation causes a relatively mild and slowly progressive myopathy. The most significant symptoms were tibialis anterior, peroneal and finger extensor muscles weakness, associated with pseudo-hypertrophy of calves and moderate quadriceps weakness and atrophy. This clinical phenotype specially characterised by distal upper and lower limbs involvement is peculiar and different from RYR1-related AD-CCD where proximal weakness and kyphoscoliosis are constantly observed. By contrast, the distal involvement present in our patients is coherent with other forms of MYH7-related disorders harbouring mutations in the myosin tail domain of the slow-myosin heavy chain (MYH7) gene (figure 1C).3–12 However, some significant differences were noted; notably ‘hanging big toe’, the characteristic sign of Laing myopathy, was not found in patients reported here.12
The muscle imaging of lower limbs from these AD-ECD patients showed the same abnormalities. Tibialis anterior muscles were affected early. Then, the most affected muscles were quadriceps, glutaeus medius, lateral gastrocnemius and peroneal. It is interesting to note that MRI analysis showed a marked difference of the muscle involution between patients P.II-2 and P.III-3 performed at 70 and 36 years old, respectively, which seem to demonstrate the slowly evolution of the disease (figure 2). This pattern is completely different from that found in a CCD patient with heterozygous RYR1 mutations.13
In this family, there was a missense p.Leu1723Pro mutation in the MYH7 gene, affecting a residue located in the mid-region of the rod domain of slow/β cardiac MyHC. It is noticeable that mutations in the mid and distal portion of rod domain of MYH7 lead to different clinical and morphological phenotypes.4–9 ,12
In conclusion, our observation indicates that AD-ECD without cardiac involvement is a clinico/pathological phenotype associated with the MYH7-related disorders, allowing us to enlarge the genetic spectrum of AD-CCD myopathies. MYH7 should be considered as a cause of AD-congenital myopathies with cores not related to RYR1.
Acknowledgments
We are grateful to patients attending the Institute of Myology (Paris) and the Neurology service (Poitiers). We would like to thank G Brochier, MT Viou, M Beuvin, A Madelaine and F Levy-Borsato (Institut de Myologie, Unité de Morphologie Neuromusculaire) for their excellent technical assistance.
Footnotes
-
Contributors NBR: Study design, performed clinical and muscle biopsies data and analysis, and wrote the manuscript. TX, US: Performed genetic data analysis. EM: Performed clinical data analysis and wrote the manuscript. JB, BW, FX: Contributed genetic data analysis. SB: Contributed muscle MRI analysis. SM, J-PN: Contributed clinical data. NM: Performed RYR1 study exclusion. MF: Performed clinical and muscle pathology data and wrote the manuscript. JL: Study design, performed genetic data analysis and wrote the manuscript. All authors read the manuscript.
-
Funding This work was supported by the Agence Nationale de la Recherche (ANR-11-BSV1-026) and the Assistance Publique-Hôpitaux de Paris (AP-HP), the Institut National de la Santé et de la Recherche Médicale (INSERM), Centre National de la Recherche Scientifique (CNRS), Strasbourg University, Collège de France, Association Française contre les Myopathies (AFM).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Ethical committee La Pitié-Salpêtrière Hospital.
-
Provenance and peer review Not commissioned; externally peer reviewed.