Article Text

Abstract
Objective To examine the long-term impact of early treatment initiation of interferon beta-1b (IFNB1b, Betaferon/Betaseron) in patients with a first event suggestive of multiple sclerosis (MS).
Methods In the original placebo-controlled phase of BENEFIT, patients were randomised to IFNB1b 250 μg or placebo subcutaneously every other day. After 2 years or diagnosis of clinically definite MS (CDMS), all patients were offered open-label IFNB1b treatment for a maximum duration of 5 years. Thereafter, patients were enrolled in an observational extension study for up to 8.7 years.
Results Of the initial 468 patients, 284 (60.7%; IFNB1b: 178 (61.0% of the original arm), placebo: 106 (60.2% of original arm)) were enrolled in the extension study. 94.2% of patients were receiving IFNB1b. Patients originally randomised to IFNB1b had a reduced risk of developing CDMS by 32.2% over the 8-year observation period (HR 0.678; 95% CI 0.525 to 0.875; p=0.0030), a longer median time to CDMS by 1345 days (95% CI 389 to 2301), and a lower annualised relapse rate (0.196 (95% CI 0.176 to 0.218) versus 0.255 (95% CI 0.226 to 0.287), p=0.0012), with differences mainly emerging in the first year of the study. Cognitive outcomes remained higher in the early treated patients. EDSS remained low over time with a median of 1.5 in both arms.
Conclusions These 8-year results provide further evidence supporting early initiation of treatment with IFNB1b in patients with a first event suggestive of MS.
- MULTIPLE SCLEROSIS
- INTERFERON
- INTERVENTIONAL
- RANDOMISED TRIALS
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system that can lead to the accumulation of significant disability.1 As the disease typically lasts for several decades, long-term follow-up studies are important to better understand the impact of disease-modifying therapies (DMTs). However, conducting these trials can be challenging due to many methodological barriers, including the difficulties associated with patient ascertainment.2
Clinical trials have shown that the early initiation of treatment with interferon beta-1b (IFNB1b; Betaferon/Betaseron; Bayer HealthCare Pharmaceuticals) can improve outcomes for patients with MS. The BENEFIT (BEtaferon/BEtaseron in Newly Emerging MS For Initial Treatment) study examined the impact of IFNB1b in patients with a clinically isolated syndrome (CIS). In this trial, early IFNB1b treatment reduced the number of patients progressing to clinically definite MS (CDMS) compared with placebo after 3 years (37% vs 51%, hazard ratios (HR) 0.59) and 5 years (46% vs 57%, HR 0.63).3 ,4 Annualised relapse rate (ARR) and Paced Auditory Serial Addition Task (PASAT) scores were also in favour of the early treatment arm.3 ,4
We report results of an 8-year, non-interventional extension of BENEFIT focusing on the description of the course of disease in the overall study population and on the comparison of outcomes in the early versus delayed treatment arms.
Methods
Patients and procedures
The BENEFIT study comprised a prospective, 2-year, international, multicentre, randomised, double-blind, placebo-controlled, parallel-group, phase 3 study with a preplanned follow-up phase of up to 5 years, followed by an observational extension study (NCT00544037) for a maximum follow-up of 8.7 years. The protocol was approved by the institutional review boards of the participating institutions, and informed consent was collected at enrolment into each phase.3–5 Eligible patients had experienced a first neurological event suggestive of MS (CIS) and had at least two clinically silent lesions on T2-weighted brain MRI. Within 60 days, patients were randomly assigned in a 5:3 ratio to IFNB1b 250 µg or placebo subcutaneously every other day. Patients completed the placebo-controlled phase when CDMS was diagnosed using Poser criteria6 or after 2 years in the study (whichever came first) and were then eligible to enter the open-label, single-arm, follow-up study for up to 5 years during which all patients were offered IFNB1b, but could also opt to take other or no medication. Blinding of the initial randomisation was maintained throughout the 5-year period. Prospectively planned integrated analyses were performed at 3 5 years after the first event suggestive of MS.3 ,4
All 468 patients randomised and treated at least once with study medication in the placebo-controlled phase were eligible to enter the observational extension study in which treatment decisions were made exclusively at the discretion of physicians and patients. Physicians recorded any medications for MS that were administered. Prior to the end of the BENEFIT Extension study, the steering committee identified a list of therapies that were defined as DMTs. Of this list, a subset was considered as escalation therapy (figure 1).

Proportion of patients requiring escalation therapy pooled across total population. The majority of patients did not require escalation therapy during the study. The treatments used by the 6.6% who required escalation therapy are shown in the inset box. Escalation therapies included alemtuzumab, cyclosporine, cladribine, cyclophosphamide, daclizumab, fingolimod, fingolimod hydrochloride, methotrexate, methotrexate sodium, mitoxantrone, mitoxantrone hydrochloride, mycophenolate mofetil, mycophenolate sodium, natalizumab, rituximab, sirolimus, tacrolimus and temsirolimus. aWhen multiple therapies are listed, the order indicates the sequence of therapies. Inset box lists only the escalation therapies administered during the study. Escalation therapy was generally similar between groups, with the exception of natalizumab (early treatment, 10 patients (3.4%); delayed treatment, 9 patients (5.1%)).
Evaluations were performed at baseline and every 6 months until December 2010. MRI outcomes, including time to McDonald MS, could not be sufficiently assessed in the observational extension study. No serum samples were taken in the extension phase. Neutralising antibodies (NAbs) were thoroughly assessed in the 5-year interventional follow-up phase.7
In addition to the original key outcome measures time to CDMS, ARR, and sustained Expanded Disability Status Scale (EDSS) score,3 ,4 time to first escalation therapy was included as an outcome in the extension study. Sustained EDSS progression was defined as an increase by ≥1 point at two consecutive scheduled visits (at least 140 days apart) and maintained throughout all subsequent scheduled or unscheduled visits in the study as compared with the lowest EDSS score during screening or baseline. Scores obtained during relapses were included. Additional outcome measures included the MS Functional Composite (MSFC),8 conversion to Secondary Progressive MS, and patient-reported quality of life (QoL) as measured by the Functional Assessment of MS (FAMS), the FAMS-Trial Outcome Index (FAMS-TOI)9 and the EuroQoL 5-Dimensional questionnaire (EQ-5D).10
Statistical analyses
Statistical analyses were conducted on the dataset of the entire BENEFIT study period. All variables were analysed by descriptive statistical methods. Efficacy was analysed in three domains: relapse-based, disability-based and patient-reported outcome-based variables (see online supplemental appendix 2). Most variables in the first domain were time-to-event variables described by Kaplan–Meier estimates, and treatment groups were compared by a log-rank test and proportional hazards regression (covariates: randomised treatment, steroid use during the first clinical event, type of disease onset and categorised number of T2 lesions on BENEFIT screening MRI). The variables in the last two domains were mainly analysed using non-parametric and/or parametric longitudinal modelling. Predefined statistical modelling procedures were used to estimate treatment effects and explore the relationship of target variables to treatment and prognostic covariates. Use of DMTs was analysed by descriptive statistical methods.
Role of the funding source
The study was designed by members of the steering committee and the study sponsor. The authors had access to all the data, participated in analysis and interpretation, and were members of the publication committee. The decision to submit the article for publication was made jointly by the members of the steering committee.
Results
Patient disposition and demographics
Of the 468 patients originally randomised in the BENEFIT study (IFNB1b: 292; placebo: 176),6 284 patients (60.7%) were enrolled in the extension phase (178 patients (61.0%) originally randomised to IFNB1b (early treatment group) and 106 patients (60.2%) originally randomised to placebo (delayed treatment group), see online supplemental figure S1). These patients were recruited from 72 of the 97 initial centres in 17 of the 20 initial countries. Excluding patients from non-participating sites (which is likely to be a random rather than a selection bias), the enrolment rate was 67.8%. Fifty-five per cent of patients (including all sites) completed the extension study (61.6% excluding non-participating sites). Baseline characteristics were generally similar across the two groups on entry to the randomised and extension phases (table 1). Patients who entered the extension study after 5 years had experienced a higher rate of conversion to CDMS (50.7% vs 40.2%) and McDonald MS (85.2% vs 74.5%) than patients who did not enter the extension phase.
Baseline characteristics of patients who did and did not enter the BENEFIT Extension study (A) at the start of the placebo-controlled phase of BENEFIT and (B) at the start of the extension study
DMT and escalation therapy usage
Of the original 468 patients, 441 (94.2%) received IFNB1b since the start of BENEFIT. The median relative time (MRT) on study drug (the time on study drug relative to the time under observation) was 75.2% among patients taking IFNB1b at least once. For 363 patients (77.6%), IFNB1b was the only DMT recorded at any time. More than half the patients (55.6%) in the overall study population were still on IFNB1b within the 90-day period before their last study day. Other DMTs administered at some point over the 8-year observation period included interferon beta-1a (Avonex, Biogen Idec; 28 patients (6.0%), MRT 19.0%) and glatiramer acetate (Copaxone, Teva Neuroscience; 27 patients (5.8%), MRT 23.8%). In the delayed treatment arm, the length of placebo exposure was a minimum of 1 month and a median of 23.0 months (mean 17.5 months). Only 24 patients (5.1%) received no DMT. Eighty-three percent (142/171) of patients taking IFNB1b at least once in the extension study were still on this treatment within the 90-day period before their last study day.
Overall, the percentage of patients who underwent escalation therapy remained low. Only 31 patients (6.6%) received other DMTs that were considered escalation therapies over the course of the 8-year study, in most cases natalizumab (figure 1). Sixteen of these 31 patients subsequently stopped their first escalation therapy within the study period; four subsequently used ≥1 therapy. At the end of the study period, the probability of patients requiring first escalation therapy remained low and was similar in both the early and delayed treatment groups (Kaplan–Meier estimates 9.3% vs 10.7% as of year 8). Among the patients who received escalation therapy, the ARR was 0.617 (95% CI 0.518 to 0.729) versus 0.185 (95% CI 0.169 to 0.202) in those who did not receive therapeutic escalation. The last available EDSS value prior to the patient's first therapeutic escalation was higher (median 2.5) than in the whole study population.
Study outcomes
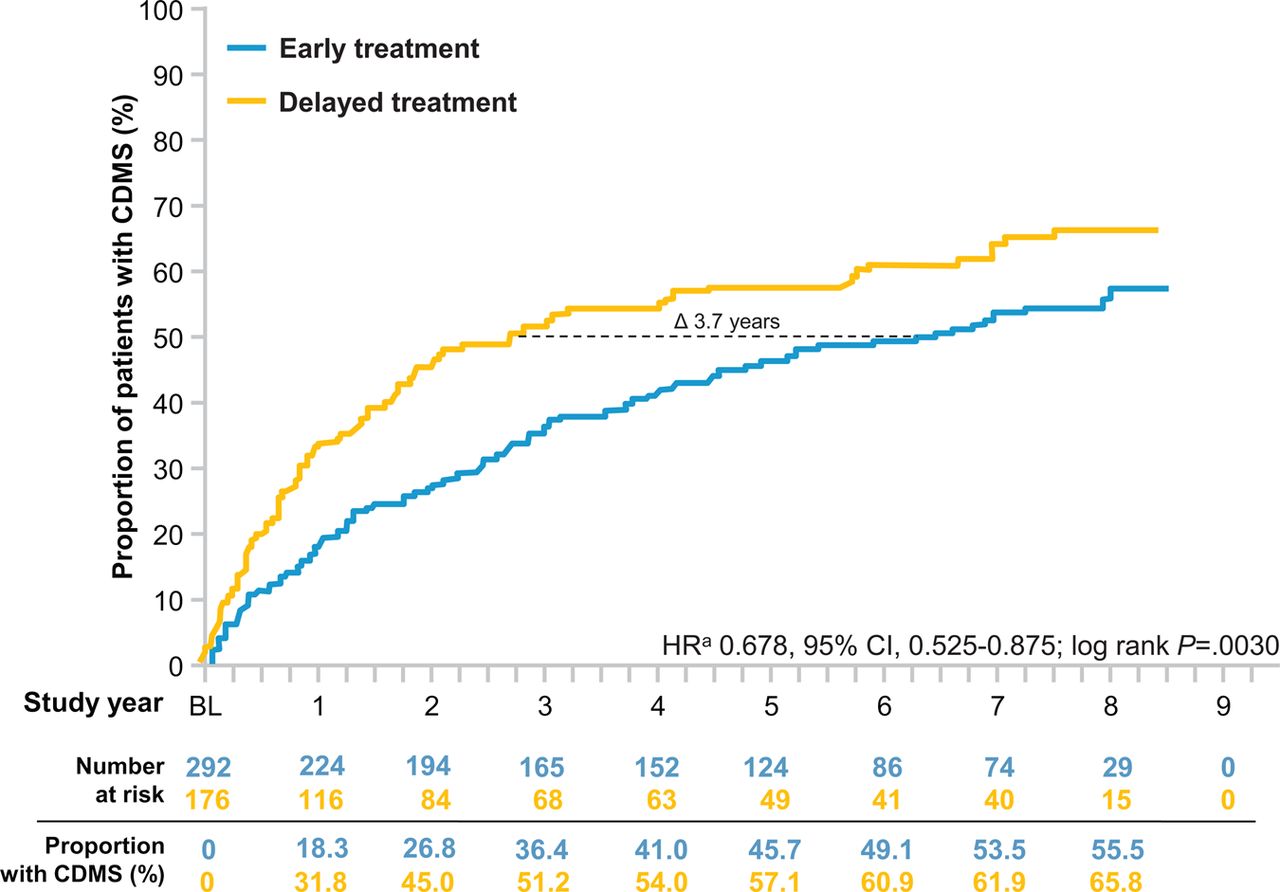
At the end of the 8-year observational period, the risk for development of CDMS in the early treatment group was lower than that of the delayed treatment group by 32.2% (HR 0.678, 95% CI 0.525 to 0.875; p=0.0030, log-rank test; figure 2). Based on Kaplan–Meier estimates, early treatment with IFNB1b reduced the probability of the development of CDMS over the 8 years (55.5% early treatment vs 65.8% delayed) with differences emerging between the treatment arms in the first year. At the 50th percentile, IFNB1b prolonged the time to CDMS by 1345 days (3.7 years, 95% CI 389 to 2301; 2335 days (6.5 years) in the early versus 990 days (2.8 years) in the delayed treatment group.

Kaplan–Meier estimates for the probability of CDMS over 8 years. Probability of conversion to CDMS was significantly higher in the delayed treatment group. At the 50th percentile, conversion to CDMS was delayed by approximately 3.7 years in the early treatment group. aBy proportional hazards regression, adjusted for steroid use during the first clinical event, type of disease onset, and categorised number of T2 lesions on BENEFIT screening MRI. CDMS, clinically definite multiple sclerosis.
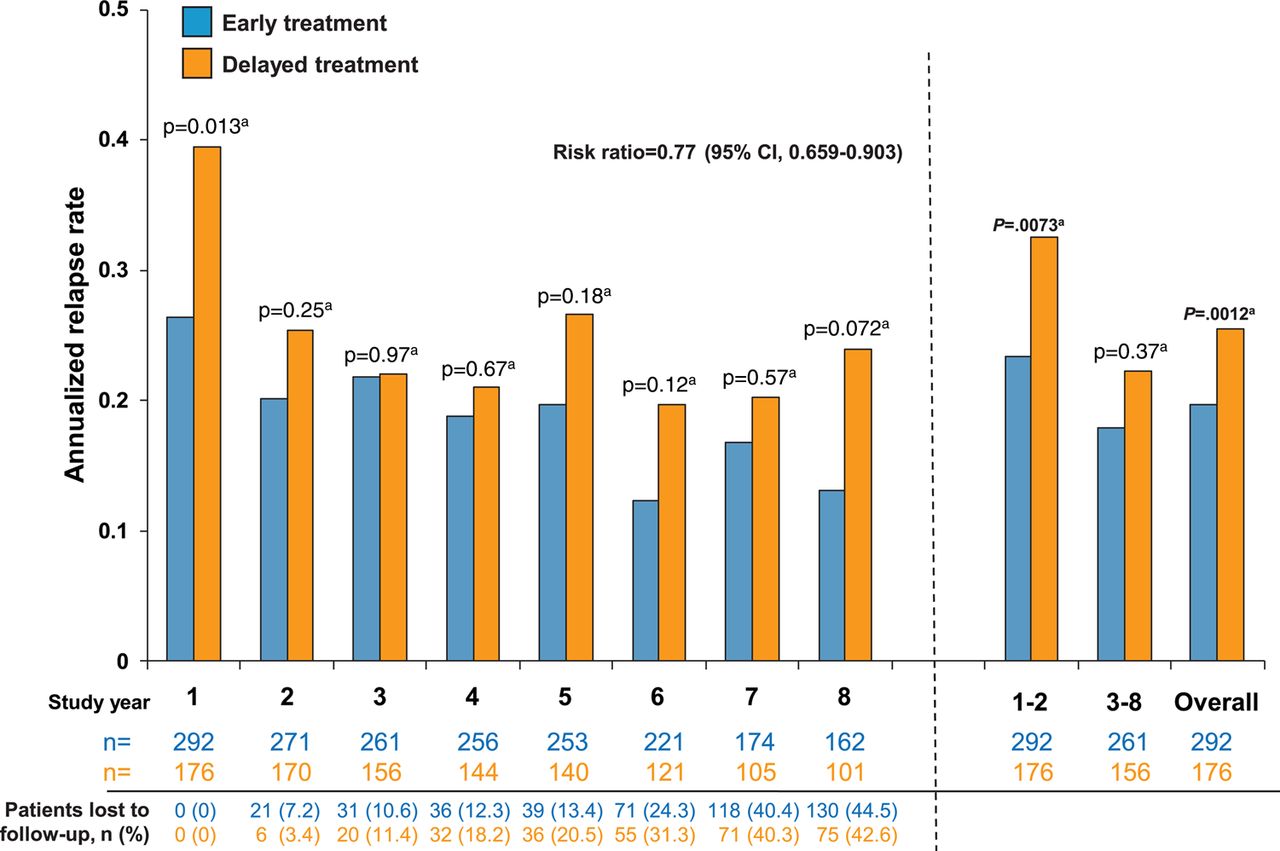
The overall ARR in the 8-year observational period was lower in the early treatment group than in the delayed treatment group (0.196 (95% CI 0.176 to 0.218) versus 0.255 (95% CI 0.226 to 0.287); figure 3). According to the generalised linear Poisson regression model, early treatment with IFNB1b reduced the ARR by 22.9% over the 8-year period compared with delayed treatment (risk ratio=0.771 (95% CI 0.659 to 0.903); p=0.0012, Wald-type χ2 test). Additionally, risk of recurrent relapses was reduced by 23.4% with early treatment (HR 0.766, 95% CI 0.589 to 0.998; p=0.048). In a post hoc analysis of ARR data, differences between treatment arms emerged in the first year of the study, with significant differences seen in combined year 1 and 2 relapse rates (early treatment: 0.233 (95% CI 0.195 to 0.277) versus delayed treatment: 0.325 (95% CI 0.268 to 0.392); p=0.0073; Wald-type χ2 test from generalised linear Poisson regression model; RR=0.705, 95% CI 0.546 to 0.910). This difference was maintained when aggregated relapse rates from years 3 to 8 were analysed (early treatment: 0.180 (95% CI 0.157 to 0.205) versus delayed treatment: 0.223 (95% CI 0.190 to 0.260); p=0.037; RR=0.807, 95% CI 0.660 to 0.987).

{kind=link}
{kind=link}
{kind=link}
Annualised relapse rate (ARR) by year in the total number of patients observed overall and during each study year. ARR was significantly lower in the early treatment group than in the delayed treatment group over the entire study period as well as in years 1 and 8. ARR was lower among patients in the early treatment than the delayed treatment group for years 1 and 2 (0.233 (95% CI 0.195 to 0.277) vs 0.325 (95% CI 0.268 to 0.392), p=0.0073a; RR=0.705, 95% CI 0.546 to 0.910) and from years 6 to 8 (0.146 (95% CI 0.115 to 0.184)) vs 0.212 (95% CI 0.164 to 0.270); p=0.032a; RR=0.694, 95% CI 0.497 to 0.969). aWald-type χ2 test. Generalised linear Poisson regression model.
There was little change from mean EDSS score at baseline/screening over the 8-year observation period, and scores were similar between the treatment groups (table 2). In both groups, the median EDSS remained 1.5 from baseline to 8 years. EDSS was stable or improved from baseline to last visit during the extension in 68.3% of patients and was worse in 31.7%. Non-parametric longitudinal modelling found no effects of time (p=0.28) or treatment (p=0.9008) on EDSS scores.
EDSS and treatment exposure
No significant differences between treatment groups, and a slight overall decrease (p=0.027) in MSFC mean z-scores including the subscores for the 9-hole peg test and 25-foot walk, were observed over the 8-year study period, although a significant increase (p=0.0453) in PASAT z-score in favour of early treatment was maintained over the whole study period (see online supplemental figure S2). Throughout the 8-year observational period, QoL as measured by FAMS-TOI (see online supplemental figure S3) and EQ-5D rating scales remained high, without significant differences between the two treatment groups (data not shown).
Neutralising antibodies
Only 39 (n=25 (14.1%) IFNB1b arm; n=14 (13.2%) placebo arm) of the patients enrolled in BENEFIT Extension were NAb+ at their last assessment in the interventional follow-up phase of BENEFIT. When data were stratified into four groups (NAb:- <20 NU/mL, NAb+: ≥20 to <100 NU/mL, ≥100 to <400 NU/mL, ≥400 NU/mL), no differences in EDSS or conversion to CDMS were identified. Median EDSS was 1.5, 1.5, 2.0 and 1.5, respectively, in these groups at their last visit in the extension study. Thus, descriptive analyses neither showed an increased rate of conversion to CDMS nor EDSS progression during the non-interventional extension period in patients who were NAb+ at the end of the interventional study.
Safety and tolerability results
In the extension study, the frequency of adverse events (AEs) was within the well-established safety and tolerability profile for IFNB1b; there were no new unexpected safety signals. The total number of patients experiencing ≥1 serious AEs (SAE) in the extension study was similar in each group: 12 patients (6.7%) in the early treatment group and eight patients (7.5%) in the delayed treatment group. Almost all SAEs were reported to be unrelated to IFNB1b, as assessed by the investigators.
Discussion
In this observational extension of the BENEFIT trial, patient ascertainment remained modestly high after 8 years with little difference in baseline characteristics when participating patients were compared with those not entering the extension. Overall, patients in both the early and the delayed treatment groups who entered the BENEFIT Extension tended to have had more active disease during the double-blind study, as depicted by higher rates of CDMS and McDonald MS. This would be expected to reduce the differences between treatment groups. However, significant differences between early and delayed treatment were still observed.
Patients who received early treatment had a lower risk of conversion to CDMS, lower ARR and longer time to recurrent relapses than those who received delayed treatment, differences that persisted throughout the study. After 8 years, 51.9% (45.5% by Kaplan–Meier estimates) of the patients who received early treatment in BENEFIT had not converted to CDMS, which contrasts with the subgroup of the CIS cohort reported by Fisniku et al11 with ≥1 clinically silent MRI lesion at screening in which only 17% had not converted to CDMS after 9.7 years. Although the differences in outcomes appear to be mainly driven by treatment effects that emerged in the first year of the BENEFIT study and would be expected to dissipate as patients in the delayed treatment arm accumulated more time on IFNB1b, earlier treatment seemed to have continued benefits over the long term. For example, although differences in ARR across treatment groups were mainly driven by treatment effects in the first year of the study, a significant difference between treatment arms was still found across years 3–8, which might indicate a prolonged benefit due to an immunomodulatory shift that cannot be recovered in the delayed treatment group.
Early treatment also appeared to provide some cognitive benefits although some longitudinal variation in PASAT scores was observed, potentially due to practice effects and changes in the study population over time. The improved cognitive performance in patients who underwent early treatment was consistent with earlier analyses from BENEFIT.4 ,12 PASAT results suggest that minimising inflammation through treatment with IFNB1b in the early stages of the disease may preserve cognitive capacity of patients with MS.
By contrast with these endpoints where the treatment arms displayed differences, EDSS, MSFC total z-score, time to first escalation therapy, FAMS-TOI and EQ-5D scores did not differ between the groups, but generally showed little change over the course of the study. In general, disease activity remained low for most patients over the 8-year period of the study in both groups. Median EDSS scores at year 8 were 1.5 and only 13.7% of patients participating in the Extension study reached sustained EDSS 3 at any time during the 8-year period. For comparison, in a natural history cohort of patients with relapsing-remitting MS published by Scalfari et al13, and in a more contemporary partially treated cohort reported by Leray et al14 (with a mean time between clinical onset and treatment start of 7.4 years), the median time to sustained EDSS 3 was 10 years from disease onset.14 The number of patients in BENEFIT reaching EDSS 3 thus seems smaller than could be expected, suggesting an impact of early treatment on disability progression. The observed long-term disease stability was also reflected in the low number of patients requiring escalation therapy in both arms. NAb status measured at the last visit of the interventional trial period did not result in any discernible differences in clinical outcomes. Patients who were NAb− at that time would be unlikely to have converted to NAb+ in the extension study.
Although the terms early and delayed treatment have been applied to the different treatment groups in this analysis of the BENEFIT study, it should be noted that both groups of patients started treatment within the first 2 years from CIS (eg, relatively early in the course of their disease). Consequently, there were only minor differences in treatment exposure between these groups. Despite the relatively small difference in time on treatment, benefits of early treatment on ARR and disease conversion were observed with up to 8.7 years of follow-up.
Long-term retention in BENEFIT was relatively high, supporting the generally favourable tolerability of treatment with IFNB1b. No new safety signals were detected. FAMS-TOI and EQ-5D scores were relatively high and stable during the 8 years of follow-up, suggesting that medication allowed the majority of patients to maintain QoL.
The findings from this study are largely in agreement with those of the Controlled High Risk Avonex MS Prevention Study in Ongoing Neurological Surveillance (CHAMPIONS) study of the efficacy of intramuscular interferon beta-1a for patients with a CIS.15 ,16 With long-term follow-up (8 years in BENEFIT, 10 years in CHAMPIONS) both trials reported comparable effects of early treatment on rates of CDMS progression and ARR, with most patients also showing little or no EDSS progression. However, there were some methodological differences between CHAMPIONS and BENEFIT, including differences in the length of blinding and preplanned follow-up (3 vs 5 years in both cases).15 Additionally, the BENEFIT population was slightly different from the CHAMPIONS population, all of whom were treated with corticosteroids at CIS (vs 70.9% in BENEFIT), and who tended to be older (median of 34 vs 30 years at baseline in BENEFIT).16 ,17 Overall, extension study enrolment was lower in CHAMPIONS (40.5% compared with 60.7% in BENEFIT).16 Last, CHAMPIONS did not report cognitive outcomes.16
Key factors of the present study that are critical to assessing its validity include the potential for ascertainment bias in the study population, differences in medication exposure across treatment arms, and the potential for gaps in patient observation. Compared with other studies of CIS,16 ,18 ascertainment in the BENEFIT Extension study was relatively high. Additionally, differences in baseline characteristics were minimal, suggesting limited selection bias. By contrast, on-study outcomes after 5 years showed that patients with more active disease entered the extension, which may have led to an overestimation of disease progression in the delayed treatment and early treatment groups. Finally, varying treatments, drug holidays and lack of adherence during the observational phase should have lessened any treatment effects and biased the results toward the null hypothesis of no difference between treatment groups. Despite this possibility, a consistent benefit of early treatment was identified.
This extension study showed a low overall disease activity and progression rate in patients with CIS. Regarding conversion to CDMS and relapse frequency, early treatment retained significant benefits as compared with delayed treatment. These findings and the benign long-term safety profile of IFNB1b, support the early initiation of such treatment in patients with CIS. The observed low EDSS progression and relapse rates in BENEFIT—both regarded as positive long-term predictors13—give hope that early treatment might have a positive influence on long-term outcomes beyond the first 8 years of treatment. In order to better understand the chronic course of this disease, we remain committed to continued follow-up of this patient cohort, including further assessment of imaging outcomes.
Acknowledgments
We are grateful to the patients and the BENEFIT investigators for their continuing contributions to the study. Additionally, we thank Charlotte Stolz (PAREXEL International GmbH) for statistical planning and programming support. Robert C Ristuccia, PhD, provided medical writing assistance in preparing the manuscript (Precept Medical Communications) that was funded by Bayer HealthCare Pharmaceuticals.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online appendix
Footnotes
GE, LK, RS and DP contributed equally to this study.
-
The Benefit study group members are listed in appendix 1.
-
Collaborators See online supplemental appendix 1 for the members of the group.
-
Contributors GE: Interpreted data and actively contributed to the writing and reviewing of the submitted manuscript. Member of the study steering committee and study investigator. LK: Interpreted data and actively contributed to the writing and reviewing of the submitted manuscript. Member of the study steering committee and study investigator. XM: Interpreted data and actively contributed to the writing and reviewing of the submitted manuscript. Member of the study steering committee and study investigator. CHP: Interpreted data and actively contributed to the writing and reviewing of the submitted manuscript. Member of the study steering committee and study investigator. MSF: Interpreted data and actively contributed to the writing and reviewing of the submitted manuscript. Member of the study steering committee and study investigator. H-PH: Interpreted data and actively contributed to the writing and reviewing of the submitted manuscript. Member of the study steering committee and study investigator. DM: Interpreted data and actively contributed to the writing and reviewing of the submitted manuscript. Member of the study steering committee and study investigator. FB: Collected and analysed the MRI data, reviewed the statistical analyses, and actively contributed to the writing and reviewing of the submitted manuscript. JH: Developed the statistical analysis plan, conducted the statistical analysis, interpreted data, and actively contributed to the writing and reviewing of the manuscript. VL: Developed the observational plan/protocol and statistical analysis plan, interpreted data, and actively contributed to the reviewing of the manuscript. BS: Drafted the statistical analysis plan, interpreted data, and drafted and reviewed the manuscript. CP: Actively involved in drafting the MRI protocol and the statistical analysis plan, reviewed the statistical analyses, and actively contributed to the writing and reviewing of manuscript drafts. Sponsor's responsible clinician for the follow-up study phase. RS: Interpreted data and actively contributed to the writing and reviewing of the submitted manuscript. Member of the study steering committee. Sponsor's responsible clinician for the placebo-controlled study phase. DP: Drafted the statistical analysis plan, interpreted data and drafted and reviewed the manuscript. Sponsor's responsible clinician for the extension study phase.
-
Funding This study was funded by Bayer HealthCare Pharmaceuticals Inc.
-
Competing interests GE has received honoraria for lectures or consulting from Biogen Idec, Merck Serono and Sanofi Aventis, received personal compensation for serving on the BENEFIT scientific advisory board, and for speaking from Bayer Schering Pharma AG. GE has also received research support from Serono: grant to University Hospital to support a research programme on MRI in MS and from Teva: grant to support a research programme on anti-IBF neutralising antibodies; LK has lectured at medical conferences or in public and received honoraria for participation in advisory boards and steering committees which have been exclusively used for funding research for his department. Research and the clinical operations (nursing and patient care services) of the MS Center in Basel have been supported by non-restricted grants from Actelion Pharmaceuticals Ltd, Advancell, Allozyne, BaroFold, Bayer HealthCare Pharmaceuticals, Bayer Schering Pharma, Bayhill, Biogen Idec, Biotica, Elan, Genmab, GeNeuro SA, GlaxoSmithKline, Mitsubishi Pharma, Merck Serono, MediciNova, Novartis, Novonordisk, Octapharma, Peptimmune, Roche, Sanofi Aventis, Santhera Pharmaceuticals, Teva, Xenoport, UCB, Wyeth and by grants from the Swiss MS Society, the Swiss National Research Foundation, the European Union, the Gianni Rubatto Foundation, Novartis, and Roche Research Foundations. XM has received speaking honoraria and travel expenses for scientific meetings, has been a steering committee member of clinical trials or participated in advisory boards of clinical trials in the past years with Bayer, Biogen Idec, EMD, Genentech, Genzyme, Merck Serono, Neurotec, Novartis, Sanofi Aventis, Teva Pharmaceuticals and Almirall; CHP has received personal compensation from Biogen Idec, Bayer Schering Pharma AG, Teva Pharmaceutical Industries, Merck Serono, MorphoSys, Novartis Pharmaceutical Corporation, GlaxoSmithKline, Actelion Pharmaceuticals Ltd, UCB, Receptos, and Roche for consulting services. The VU University Medical Center received financial support for research activities from Bayer Schering, Biogen Idec, Merck Serono, Teva, Novartis, GSK, and Roche; MSF has received compensation from Actelion, BayerHealthcare, Biogen Idec, Celgene, EMD Canada, Genzyme, Glycominds, Hoffman La-Roche, Merck Serono, Novartis, Opexa, Sanofi Aventis, Teva Canada Innovation for consulting services and has received research/educational grants from Bayer HealthCare and Genzyme. He also participates in a Genzyme-sponsored speaker's bureau; H-PH has received honoraria for consulting and speaking at symposia from Bayer-Schering Pharma, Biogen Idec, Genzyme, Merck Serono, Novartis, Roche, Teva, and Sanofi Aventis, with approval by the rector of Heinrich-Heine University; DM has received honoraria through payments to his employer, UCL Institute of Neurology, for Advisory Committee and/or Consultancy advice in multiple sclerosis studies from Biogen Idec, GlaxoSmithKline, Novartis, Merck, Chugai, Mitsubishi Pharma Europe, and Bayer Schering Pharma. DM has also received compensation through payments to his employer for performing central MRI analysis of multiple sclerosis trials from GlaxoSmithKline, Biogen Idec, Novartis and Merck. The NMR Research Unit at UCL Institute of Neurology is supported by the UK MS Society and UCL-UCLH Biomedical Research Centre; FB has received compensation for consultancy from Bayer-Schering Pharma, Biogen Idec, Merck Serono, Novartis, Sanofi Aventis, Genzyme, Roche, Teva and has received research support from the Dutch Foundation for MS research (an NGO); JH is a salaried employee of PAREXEL International; VL is a salaried employee of Bayer Pharma AG; BS is a salaried employee of Bayer Pharma AG/Bayer HealthCare Pharmaceuticals; CP is a salaried employee of Bayer Pharma AG/Bayer HealthCare Pharmaceuticals. CP owns stock in Bayer AG, the owner of Bayer Pharma AG/Bayer HealthCare Pharmaceuticals; RS is a salaried employee of Bayer Pharma AG/Bayer HealthCare Pharmaceuticals. RS owns stock in Bayer AG, the owner of Bayer Pharma AG/Bayer HealthCare Pharmaceuticals; DP is a salaried employee of Myelo Therapeutics GmbH. DP is a former salaried employee and is currently a paid consultant for Bayer Pharma AG/Bayer HealthCare Pharmaceuticals. DP owns stock in Bayer AG, the owner of Bayer Pharma AG/Bayer HealthCare Pharmaceuticals.
-
Ethics approval The protocol was approved by the institutional review boards of the participating institutions.
-
Provenance and peer review Not commissioned; externally peer reviewed.