Article Text

Abstract
Cerebral vasospasm has traditionally been regarded as an important cause of delayed cerebral ischaemia (DCI) which occurs after aneurysmal subarachnoid haemorrhage, and often leads to cerebral infarction and poor neurological outcome. However, data from recent studies argue against a pure focus on vasospasm as the cause of delayed ischaemic complications. Findings that marked reduction in the incidence of vasospasm does not translate to a reduction in DCI, or better outcomes has intensified research into other possible mechanisms which may promote ischaemic complications. Early brain injury and cell death, blood-brain barrier disruption and initiation of an inflammatory cascade, microvascular spasm, microthrombosis, cortical spreading depolarisations and failure of cerebral autoregulation, have all been implicated in the pathophysiology of DCI. This review summarises the current knowledge about the mechanisms underlying the development of DCI. Furthermore, it aims to describe and categorise the known pharmacological treatment options with respect to the presumed mechanism of action and its role in DCI.
- CEREBRAL BLOOD FLOW
- SUBARACHNOID HAEMORRHAGE
- CEREBROVASCULAR DISEASE
Statistics from Altmetric.com
Introduction
The incidence of spontaneous subarachnoid haemorrhage (SAH) is around 6–11 per 100 000 persons per year.1 ,2 Due to the relatively young age of the affected population and the high rates of disability, the burden to society is high, with a reported loss of productive years similar to ischaemic and haemorrhagic strokes.3
Over the past two decades, the advancement of understanding of the pathophysiology of SAH and its squelae has led to a considerable reduction in the mortality.4 Data suggest that this reduction may be related to new management protocols directed at early aneurysm repair, and aggressive management of acute hydrocephalus and delayed cerebral ischaemia (DCI).4 ,5 Despite these advances, about 30% of patients who survive following SAH will not regain full independence,6 while 69% will report a reduced quality of life.7
DCI is recognised as one of the leading causes of unfavourble outcome following SAH.8 Understanding the exact mechanisms which lead to DCI is important in the development of new treatment strategies. Furthermore, with multiple therapies being tested, it is important to understand the background behind the interventions, as well as the current state of evidence for their likely benefit.
DCI
DCI has been shown to occur in 30–40% of patients with SAH.8 ,9–11 The pathophysiology of DCI is complex and not fully understood. Until recently, the prevailing view has been held that there is a direct link between arterial narrowing seen on angiography and clinical symptoms of brain ischaemia. However, recent data argue that this relationship is inconsistent. While angiography and perfusion imaging often demonstrate vasospasm and associated perfusion deficits, this is by no means invariable, and in many cases, DCI may be a diagnosis of exclusion without clear radiological findings.
Cerebral vasospasm
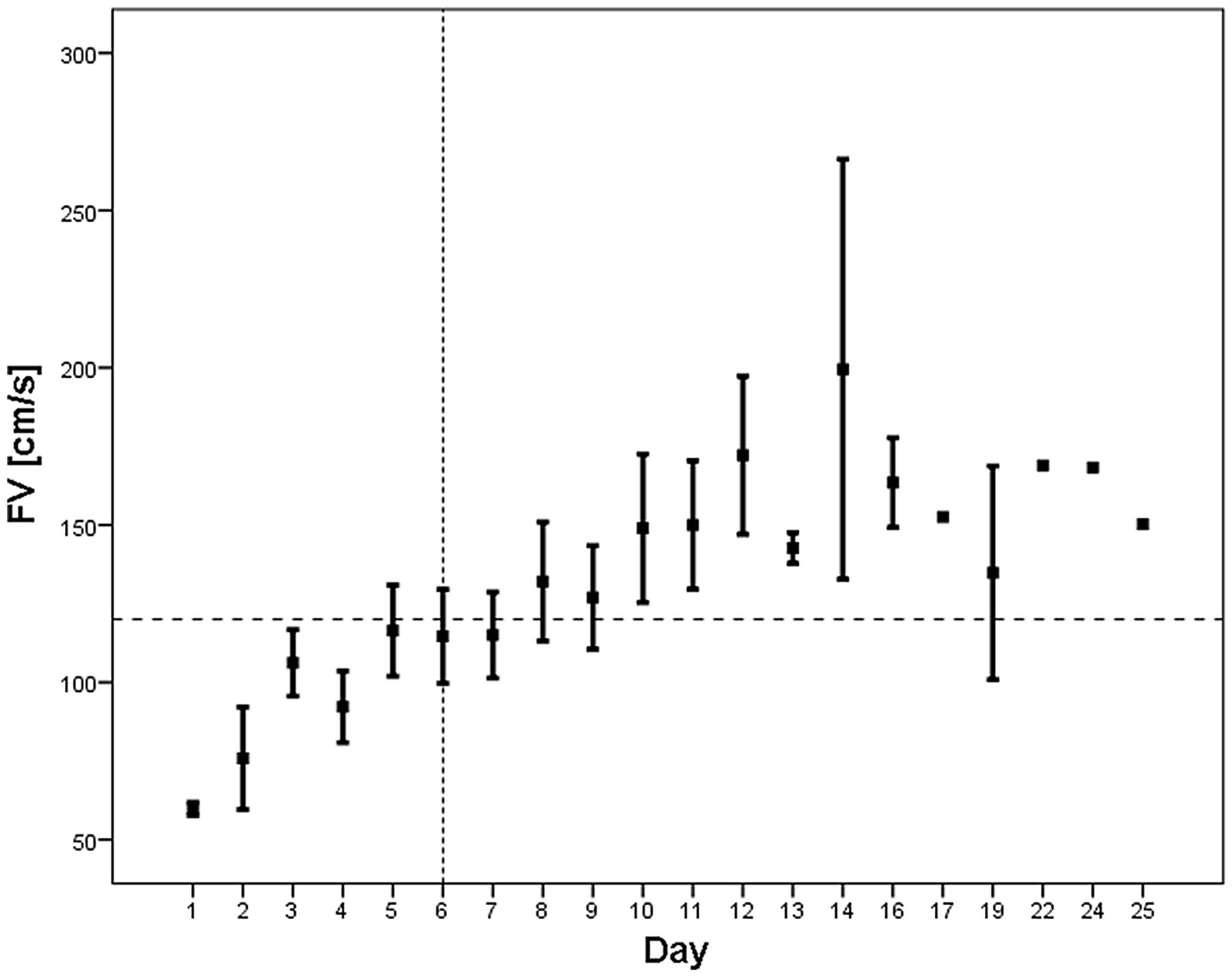
Ecker and Riemenschneider12 first documented the presence of cerebral vasospasm (CVS) in relation to a ruptured aneurysm. Allcock and Drake13 demonstrated a link between vasospasm and symptoms of focal ischaemia. Arterial narrowing has typically been shown to have a delayed onset and a peak between 5 and 14 days, following which it typically resolves (figure 1).14 It is, therefore, understandable as to why CVS has been related to delayed deterioration.14–18

Flow velocity changes in a cohort of subarachnoid haemorrhage patients. An increase of FV can be seen from day 6 with a peak at day 10–12. Spontaneous resolution not clearly seen as patient numbers decreased in the second and third week due to discharge. Horizontal dashed line represents FV threshold of 120 cm/s, typically used in the diagnosis of cerebral vasospasm (CVS). Vertical dashed line is the median time of vasospasm onset. FV, flow velocity.
Multiple signalling pathways have been implicated in the pathogenesis of arterial spasm. The principal initiating factors are thought to be blood degradation products which accumulate in the subarachnoid space and act as triggering substances for the development of endothelial dysfunction and an intramural inflammatory response.
Blood degradation products
Clinically, there is a clear link between the severity of CVS and the amount of subarachnoid blood seen on CT,19–23 a relationship recognised by the Fisher scale.19 The contractile property of cerebrospinal fluid (CSF) from patients with SAH was first described by Buckell.24 Since then it has been demonstrated that blood degradation products trigger a molecular cascade which leads to CVS. Several key observations support the role of oxyhaemoglobin, in particular, in the pathogenesis of post-SAH vasospasm. It was shown to induce vasoconstriction in cerebral arteries in vitro.25–27 Furthermore, intrathecal injection of oxyhaemoglobin, or a supernatant from autologous blood, was shown to induce vasospasm in primates. Importantly, in the same experiment, injection of methaemoglobin, bilirubin or sham CSF did not induce vasospasm. The exact mechanisms by which oxyhaemoglobin induces vasoconstriction remain unknown, but several key factors have been described. Oxyhaemoglobin is known to alter the synthesis of eicosanoids in vessel walls, in particular, decrease the production of PGI2 and increase the production of PGE2. Furthermore, oxyhaemoglobin spontaneously oxidises to methhaemoglobin releasing superoxide, which in turn, is known to lead to lipid peroxidation and vasoconstriction. Finlay, it has been demonstrated that oxyhaemoglobin inhibits endothelial-dependent relaxation.22 However, experimental and clinical studies have not, so far, demonstrated that inhibition of any one of these mechanisms alone can completely reverse vasospasm, further confirming a multidirectional effect of blood degradation products on cerebral vasculature. At present the molecular pathways remain largely unknown, and there are no effective pharmacological means to influence all the implicated mechanisms. However, approaches aimed at clearing subarachnoid spaces form blood products seem reasonable.
Inflammation
Blood-brain barrier breakdown as a consequence of SAH has been shown to lead to trafficking of lymphocytes into the CSF, as well as infiltration of arterial walls.28–31 While little direct evidence exists confirming that an induced inflammatory process may directly lead to development of vasospasm, it has been demonstrated that activated mononuclear cells within the CSF can release ET-1, a known vasoconstricting agent.32 Furthermore, blood degradation products were shown to be sufficient to induce ET-1 production by activated mononuclear cells providing a direct link between SAH, inflammation and ET-1 production.32 Longitudinal analysis of the inflammatory reaction after SAH revealed that there is a massive, compartmentalized increase in the secretion of proinflammatory cytokines such as IL-1β and IL-6.33 Furthermore, the changes in cytokine concentrations parallel the changes in blood flow velocity.33
Nitric oxide and nitric oxide synthase
Nitric oxide (NO) is one of the key endothelium-derived factors which govern vascular muscle tension. It increases 3′, 5′-cyclic guanosine monophosphate (cGMP) levels in vascular smooth muscle cells leading to vasodilatation and an increase in cerebral blood flow.34 ,35 NO levels are known to decrease following SAH in a characteristic biphasic distribution: acute—30 min after the ictus36; and delayed—around 4–7 days following the ictus.37 ,38 Whether, this is a result of binding by oxyhaemoglobin or secondary to an inflammatory process remains unknown. Furthermore, while shear stress induces vasodilatation in healthy arteries via endothelial NO synthase (NOS), this pathway is impaired following SAH,39 ,40 with a clear reduction in NOS mRNA reported on day 7 post-ictus.41 Furthermore, endogenous inhibitors of endothelial NOS, such as asymmetric dimethylargnine and protein kinase C have been described to be upregulated following SAH.42–44 Experimental and human data suggest that vasospasm can be ameliorated with the aid of exogenous NO donors such as sodium nitroprusside or nitroglycerine.45 ,46 However, adverse systemic effects of these medications (principally hypotension, which has been shown to be more pronounced than that seen with nimodipine) make them inappropriate for routine systemic administration in clinical practice. Nevertheless, it needs to be pointed that a number of studies have investigated intrathecal administration of NO donors in humans with little systemic side effects. However, the deactivation of NO exposed to oxyhaemoglobin and deoxyhaemoglobin, (formation of methaemoglobin, S-nitroso-haemoglobin and ferrous-nitrosyl-haemoglobin) remains a concern (see Pluta47 for a detailed review on NO and DCI). Recently the safety of systemic administration of sodium nitrate in humans has been confirmed, with potential clinical trials in SAH awaited.48
Endothelin-1
Endothelin-1 (ET-1), one of the most potent endogenous vasoconstrictors, produced by endothelial cells, is stimulated by ischaemic insult, but also by oxyhaemoglobin.49 The levels of ET-1 in the CSF of patients with vasospasm have been shown to be higher than those found in healthy subjects.50 ,51 Furthermore, increases in ET-1 levels correlate with the onset of ischaemic symptoms.50 ,51 However, other studies demonstrated that, although, ET-1 levels were higher in patients with DCI, they were within normal range in patients with angiographic evidence of arterial narrowing without clinical symptoms,52 suggesting that ET-1 may be a marker of ischaemic tissue damage rather than vasospasm.52 While the exact role of ET-1 in the development of CVS is unknown, it has been demonstrated that administration of ET-1 receptor type A (ETA) antagonists in an experimental setting reduced vasospasm.53 ,54 Similar findings were reported in the randomised controlled trial of Clazosentan (an ETA receptor antagonist), where inhibition of ET-1 signalling significantly reduced large vessel narrowing in a clinical setting.55–59
The relationship between vasospasm and DCI
Narrowing of cerebral arteries may cause a reduction of cerebral blood flow distal to the spastic segment, depending on the state of autoregulation, which in consequence, may lead to ischaemia and infarction.60–63 However, while up to 70% of patients demonstrate a degree of arterial narrowing on catheter angiography,64 ,65 only 20–30% develop clinical symptoms.66 The positive predictive value of vasospasm (diagnosed using stringent criteria with the aid of two imaging methods) for DCI is only 67%.8 Furthermore, up to 25% of delayed infarcts seen on follow-up CT are not located in the territory of the spastic artery, or are found in patients who did not demonstrate vasospasm.67–69
A number of studies suggested that only severe vasospasm with at least 50% luminal narrowing produces a reduction of cerebral blood flow which is sufficient to cause symptoms of ischaemia.17 ,63 ,70–74 However, others have reported that only 50% of patients with severe CVS on angiography become symptomatic.60 Positron emission tomography (PET) has shown that delayed neurological deficits after SAH were associated with a wide range of haemodynamic disturbances, ranging from hypoperfusion to hyperaemia,75 and that the spatial distribution of the haemodynamic disturbances did not always coincide with the vascular territory where narrowing was identified.70 ,75 ,76 With a more widespread use of perfusion imaging methods (such as perfusion CT) for the evaluation of DCI, similar findings are being increasingly reported (figure 2).77

(A) Cerebral angiogram of a patient with WFNS 1, Fisher 3 SAH from a ruptured left PComA aneurysm (left ICA injection) performed on day 6 postictus, demonstrates diffuse severe vasospasm in the left ICA, ACA and MCA. (B) Perfusion CT performed same day shows mild reduction in CBF with (C). a compensatory increase in CBV indicative of autoregulatory vasodilatation. (D and E) CT on day 12 demonstrates delayed infarcts in the ACA and MCA territories. (F) Cerebral angiogram of a patient with WFNS 2, Fisher 3 SAH from a ruptured right AChA aneurysm (right ICA injection) performed on day 8 postictus, demonstrates segmental vasospasm in the right ICA and MCA, as well as diffuse vasospasm in ACAs bilaterally. (G and H) Perfusion CT scan performed on the same day, demonstrates a perfusion deficit only in the left ACA territory only in CBF and CBV. (I and J) Delayed CT did not demonstrate any hypodensities. ACA, anterior cerebral artery, AChA, anterior choroidal artery; CBF, cerebral blood flow; CBV, cerebral blood volume, ICA, internal carotid artery, MCA, middle cerebral artery, SAH, subarachnoid haemorrhage; WFNS, World Federation of Neurosurgical Societies.
By contrast, some studies, with rigorous angiographic control, report that indeed, there is a significant correlation between angiographic vasospasm, DCI and delayed infarctions on follow-up imaging, and that only 3% of patients with none or only mild angiographic spasm develop delayed infarcts.16 These findings spark the question, whether other factors may responsible for the observed discrepancies. It is known that transcranial Doppler (TCD) diagnosis of vasospasm is limited mainly to the anterior circulation and, in particular, to the middle cerebral artery, therefore, spasm in other vessels may be misinterpreted. Furthermore, infarction on follow-up imaging needs to be interpreted with caution in the absence of immediate postoperative imaging to rule out other, potentially iatrogenic causes. As immediate postoperative imaging is not standard practice in many centres, this is not always reported. While in most cases, SAH is promptly diagnosed and the culprit aneurysm detected and treated, there remains a population of patients with a delayed presentation with only minimal symptoms who may have been exposed to haemodynamic instability in the early phase post-bleed.
Nevertheless, a recent meta-analysis of pharmacological treatment of vasospasm and DCI demonstrated that, despite a reduction in the incidence of CVS, there was, in most cases, little or no effect on outcome.78 Similar results were reported from the trial of clazosentan (a potent ETA receptor antagonist) as well as nicardipine (potent calcium channel blocker).55 ,79 Conversely, nimodipine, which is so far, the only drug for which class I evidence exists, reduced the incidence of DCI and poor outcome by 40%, without ameliorating vasospasm.80–82
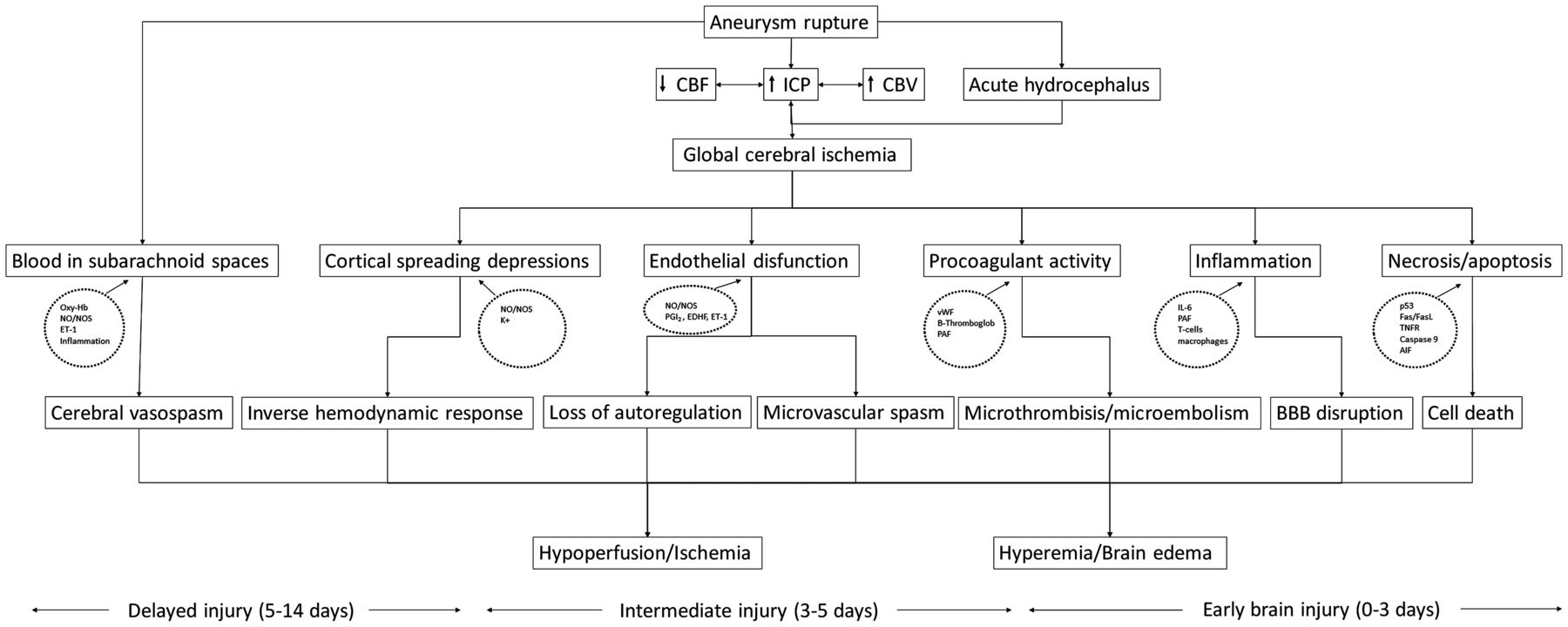
While many of the disappointing results may have been a consequence of systemic complications of the investigated compounds, which often caused blood pressure instability or pulmonary complications (such as those observed in trials of clazosentan and nicardipine), together these results argue against arterial narrowing being the sole cause of DCI. Given these findings, there is a clear need to investigate other mechanisms which may promote cerebral ischaemia following SAH. These include early brain injury (EBI), microvascular spasm, microthrombosis, spreading cortical depolarisations and failure of cerebral autoregulation (figure 3).83 ,84

Diagram depicting the possible pathophysiological pathways which may lead to development of DCI. The time ranges at bottom depict approximate/presumed periods when the various processes occur. At present, it is unknown which of the mechanisms is the main culprit, however, the paradigm is shifting away from cerebral vasospasm. AIF, apoptosis inducing factor; CBF, cerebral blood flow; CBV, cerebral blood volume; EDHF, endothelial derived hyperpolarising factor; ET-1, endothelin 1; FasL, Fas ligand; ICP, intracranial pressure; IL-6, interleukin 6; NO, nitric oxide; NOS, nitric oxide synthetase; Oxy-Hb, oxyhaemoglobin; PAF, platelet-activating factor; PGI2, prostacycline; TNFR, tumour necrosis factor receptor; vWF, von Willebrand factor; DCI, delayed cerebral ischaemia. (Based on refs. 90, 94, 95, 99).
Early brain injury
Recent reports suggest that events occurring before the onset of vasospasm, during the first 72 h after the ictus may significantly contribute to outcome following SAH.85–93 EBI includes the primary injury resulting from the ictus as well as its direct consequences.
It has been demonstrated in experimental and clinical studies that aneurysm rupture is accompanied by a severe rise of intracranial pressure, often to suprasystolic levels,94 ,95 caused by extravasation of arterial blood into the subarachnoid spaces, as well as a vasodilatory cascade.91 ,96 ,97 Intracranial hypertension leads to a decrease in cerebral perfusion pressure, and ultimately to cessation of cerebral blood flow (clinically manifested as syncope or loss of consciousness), and in consequence, global ischaemia, and later oedema.92 ,93 ,98 Another mechanism which leads to increases of intracranial pressure and reductions of cerebral blood flow is CSF outflow obstruction and acute as well as chronic hydrocephalus. Hydrocephalus may also contribute, at least partially, to the early haemodynamic disturbances and, hence, EBI.
Global cerebral ischaemia, which occurs in the acute phase of SAH, has been shown to activate several key pathophysiological pathways which, in consequence, may lead to direct nervous tissue injury as well as increased tissue vulnerability to secondary insults. These include initiation of cell death mechanisms,90 ,99 blood-brain barrier disruption,100 ,101 an acute inflammatory response,102 ,103 all of which contribute to development of cerebral oedema,104 which itself is a poor prognostic factor.85 ,105 ,106 Furthermore, the acute haemodynamic compromise may lead to microvascualr spasm63 ,107 ,108 and microthrombosis,107 ,109 ,110 as well as failure of cerebral autoregulation (figure 3).62 ,111–114 All these processes are potential players in perpetuating ischaemic injury after SAH, potentially contributing to the delayed manifestation when sufficient insults have occurred. The mechanisms implicated in DCI, along with the relevant publications are summarised in online supplementary table S1.
Oxidative stress
Experimental and clinical evidence exist supporting the role of free radicals and oxidative stress in SAH.115–122 Generation of free radicals is related to auto-oxidation of haemoglobin in the CSF, altered mitochondrial function, lipid peroxidation, as well as NADPH oxidase function.123 Studies showed that generation of free radicals is important in the pathogenesis of CVS as well as DCI.115 ,116 ,119 ,120 ,122 Human data indicates an increase in oxidative stress and lipid peroxidation in serum as well as CSF within 3 days after SAH.115 ,118 ,119 ,120 Furthermore, the increases are more pronounced in patients who developed DCI115 ,116 and those with poor neurological outcome.119 ,120 Markers of CSF lipid peroxidation peaked at day 6, suggesting a temporal relationship with DCI.115 However, due to the lack of simultaneous clinical correlation it is difficult to judge whether they precipitated of were a consequence of ischaemia. Nevertheless, in transgenic animal models it was demonstrated that an increase in the antioxidant capabilities leads to a reduction in apoptotic cell death after SAH.122
Cell death, blood-brain barrier breakdown and inflammation
Experimental data looking at cell death mechanisms is largely derived from experimental animal work due to the difficulty with available technology to image these processes in vivo. It has been demonstrated that neuronal cell death occurs within 24 h after SAH.124 ,125 Necrosis, apoptosis and autophagy have all been described in animal models, often simultaneously.126 ,127 Intrinsic, caspase-dependent pathways have been shown to be activated as early as 40 min after SAH.128 ,129 Activation of proapoptotic proteins, such as Bak, Bax, Bad and Bcl-XS is present, as well as activation of caspases 3, 8 and 9.130 ,131 On the other hand, while the early concepts and descriptions of the involved mechanisms of blood-brain barrier breakdown and neuroinflammation arise from experimental models, there have been a number of studies investigating these processes in humans using imaging and monitoring techniques. Blood-brain barrier breakdown and inflammation have also been reported in the acute phases after SAH. Animal models demonstrated that neutrophils can accumulate in cerebral vessels within 10 min after experimental SAH.132 Clinical studies looked at the major proinflammatory cytokines, for example, IL-1β, IL-6, IL-1 receptor and TNFalfa, and found that they are increased in the CSF within 3 days after SAH.133 Their increase has been associated with unfavourable outcome, vascular spasm, as well as hyperthermia.
Microvascular spasm
A study by Yundt et al134 demonstrated a diminished vasodilatory capacity of the cerebral microcirculation in patients who sustained SAH. Furthermore, data from experimental studies, where direct observations of small intraparenchymal and pial arterioles was performed, suggested the presence of microvascular spasm in two different experimental models of SAH.135–137 Ohkuma et al136 performed serial morphometric analyses of cerebral microvessels after cisternal blood injection, and demonstrated maximal luminal narrowing between days 3 and 7. Similar findings were reported in vivo in mice subjected to experimental SAH.107 In vivo microvascular spasm assessed using the cerebral circulation time on angiography showed that if present within the first 24 h, it might be predictive of subsequent large vessel vasospasm and DCI.108 Furthermore, it was demonstrated that regional reductions in cerebral blood flow are better correlated with narrowing in the microvascular compartment than with narrowing of large cerebral arteries.63 These observations argue that microvascular constriction, or the lack of microvascular dilatation may play a role in the development of DCI. The presence of microvascular spasm, not readily visible on catheter angiography, or transcranial Doppler may account for the observed discrepancies between imaging and clinical symptoms.
Microthrombosis
It has been shown that the levels of blood coagulation markers correlate with development of DCI.138 ,139 ,140 ,141 ,142 Procoagulant activity has been shown to precede DCI, with increased levels of platelet-activating factors noted on day 4 post-SAH,143 an increase in the von Willebrand factor seen as early as 72 h after the ictus,138 and loss of collagen type IV (a component of the basal lamina).109 These changes were accompanied by platelet aggregation in parenchymal vessels, which was seen as early as 10 min after experimental SAH.109 ,110 Interestingly, the timing of aggregate clearance is inconsistent with one study reporting reperfusion at 24 h,109 while in another, the peak intensity of platelet aggregation at the same time point.110 Importantly, microthrombi have also been found on autopsy of patients with SAH confirming that microthromboemboli are indeed a part of the clinical picture following SAH, in humans.144
Antifibrinolitic therapy with tranexamic acid resulted in a significant reduction in the rate of rebleeding following SAH.145–148 However, the benefit may have been counteracted by the increased incidence of DCI, which was not associated with large vessel spasm. These findings lead researchers to believe that the changes in coagulation homeostasis induced by tranexamic acid (causing microthrombosis) may have promoted DCI.149
A large systematic review and a meta-analysis performed by Darhout Mees and colleagues150 ,151 demonstrated that administration of antiplatelet medications after SAH reduces the relative risk (RR) of DCI by 15%, and shows a non-significant trend towards improved outcomes. Nevertheless, a benefit on outcome was not demonstrated, hence routine use is not recommended.
Cortical spreading depolarisation
Cortical spreading depolarisation is an abrupt electrical change with near-complete and sustained depolarisation of a neuron or group of neurons, which has a tendency to spread through the cortex, and is associated with hyperaemia. However, when clustered or affecting injured tissue cortical spreading depolarisations are associated with metabolic, biochemical and morphological dysfunction of brain parenchyma, manifested as hypoperfusion, cytotoxic oedema and hypoxia.152 ,153 Cortical spreading depressions do not normally occur in uninjured brain, however, they have been implicated in the pathophysiology of migraine. Spreading depolarisations have been observed in poor grade patients following SAH.154–156
A multicenter observational study where invasive electrocorticography was performed, the CoOperative Study on Brain Injury Depolarisations (COSBID) demonstrated that clusters of spreading depolarisation are associated, and precede, development of DCI in the absence of vascular spasm.156 The proposed mechanism responsible for propagation of DCI in these cases is thought to be an inverted haemodynamic response. In normal circumstances, a wave of spreading depolarisation is accompanied by a hyperaemic response.154 With repeated waves, this hyperaemic response is diminished, and a vasoconstrictive reaction is observed with a decreased regional cerebral blood flow and oxygen supply.154 ,157 The mechanism for the inverted haemodynamic reaction remains poorly understood. In particular, it is unclear whether the mechanism responsible for vasoconstriction and vasodilatation during waves of depolarisations are the same as for vasomotor reactions in response to chemical and pressure stimuli.
Cerebral autoregulation
Experimentally, CVS does not reduce distal cerebral blood flow unless there is an additional insult, such as a fall in blood pressure.158 This finding supports Harper's dual-control hypothesis,159–161 whereby proximal arterial spasm may be compensated by distal autoregulatory vasodilatation. However, there is a limit to such compensatory mechanisms, which is why a second insult, such as a drop in perfusion pressure or increased metabolic needs, results in insufficient blood and nutrient supply leading to ischaemia. With impaired autoregulatory mechanisms (figure 4), even a single insult, such as vessel narrowing or haemodynamic instability, may lead to significant drop in blood flow rendering the brain at an increased risk of ischaemia. Evaluation of cerebral autoregulation is being increasingly recognised as a factor requiring consideration in the management of patients with SAH.112 ,114 ,113 ,162–164 Three recent prospective studies have demonstrated that indirect indices of cerebral autoregulation can be used to prognosticate DCI as well as long-term outcome after SAH.112 ,113 ,163 ,164 ,165 Importantly, autoregulation was found to deteriorate before clinical symptoms as well as radiographically identifiable arterial narrowing.163 Whether treatment interventions can be used to alter the state of autoregulation is unknown. In a phase II randomised study of 80 patients, it was demonstrated that treatment with pravastatin shortens the duration of autoregulatory impairment.166 ,167 While a reduction in vasospasm and DCI was also observed, there was no effect on 6-month outcome.168

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Grey line depicts normal autoregulation; black line depicts different stages of impaired autoregulation, from a shift of the autoregulatory curve to complete loss of autoregulation. AR, autoregulation; CBF, cerebral blood flow; CPP, cerebral perfusion pressure.
Treatment of DCI
DCI, where insufficiency of blood and nutrient supply to the brain is present, may in consequence lead to infarction, permanent deficits and ultimately poor functional outcome. Despite the multiple mechanism involved, therapy has been largely targeting large vessel spasm. The following sections aim to summarise the current treatment strategies, for which human data is available. The available randomised controlled trials RCTs are delineated in online supplementary table 2.
Clearance of subarachnoid spaces
Blood degradation products are thought to be one of the principal mechanisms responsible for development of vasospasm,22 ,23 ,24 suggesting that rapid clearance of blood from subarachnoid spaces may have beneficial effects. Numerous methods have been investigated, including continuous cisternal drainage, lumbar drainage, as well as intrathecal administration of thrombolytics There are reports suggesting good success rates in decreasing the incidence of DCI with continuous cisternal drainage, however, a RCT has not been performed.169 Results from the first single-centre trial of early, continuous lumbar drainage hold promise, with a significant decrease in the incidence of DCI from 35% to only 21%.170 However, the study failed to demonstrate any long-term benefit in outcome. Another trial aiming to recruit 300 patients is currently ongoing (clinicaltrials.gov; NCT01258257).171
Five RCTs have evaluated the use of intrathecal thrombolytic agents to clear subarachnoid blood.172–176 A meta-analysis of these studies suggests a reduction in the incidence of DCI, as well as improvements in outcome.177 However, the benefit failed to reach statistical significance after exclusion of one study where concomitant intrathecal nimodipine was administered.174
Systemic targeting of angiographic vasospasm
Nimodipine, a calcium channel blocker, is the only drug approved for use in SAH, and is the mainstay of treatment.178 In a meta-analysis, it has been shown to reduce the risk of poor outcome, with a RR of 0.7.81 While traditionally nimodipine has been thought to prevent CVS, vascular narrowing on angiography was not included as an outcome measure in the largest trial.80 Other RCTs have demonstrated that nimodipine does not have an effect on angiographic vasospasm despite the beneficial effect on outcome, suggesting a different mechanism.82 In vitro and in vivo research demonstrates that nimodipine may have an effect on the whole vasculature, inhibiting contractions induced by noradrenaline and serotonin, potassium membrane depolarisation, as well as PGF2alfa.179 Furthermore, nimodipine has been also described to increase the fibrynolitic activity by decreasing the level of plasminogen activator inhibitor-1 (PAI-1).180
Nicardpine has been evaluated in a large, multicenter RCT in the USA—Cooperative Aneurysm Study.79 The advantage of nicardipine was the ease of preparation of the intravenous formula to be administered continuously. The study demonstrated a significant reduction of vasospasm from 46% to 32%. However, there was no benefit on outcome, with an increased number of systemic complications, such as pulmonary oedema and metabolic derangements in the treatment group.
Another vasodilatator which has been studied in SAH is fasudil. Fasudil is a potent RhoA/Rho kinase (ROCK) inhibitor, which is also thought to inhibit the action of free intracellular calcium, as well as inhibit protein kinases A, G and C, and myosin light-chain directly. Fasudil has been repeatedly shown to have beneficial effects on development of CVS, delayed cerebral infarcts as well as outcome.181 Furthermore, fasudil has been compared with nimodipine (although intravenous rather than oral) demonstrating improved outcome.182 A large multicenter study is yet to be conducted.
Cilostazol, a phosphodiesterase 3 inhibitor is a platelet aggregation inhibitor, which also has an effect on smooth muscle cells.183 Cilostazol has been shown to ameliorate vasospasm in experimental models.184 Two RCTs were conducted evaluating the use of cilostazol in SAH. One study demonstrated a benefit on DCI and outcome at discharge.185 The second study showed a reduction in the risk of vasospasm and cerebral infarction, without improvements in outcome.186 In a meta-analysis, which additionally included two non-RCT, a benefit on outcome at discharge was demonstrated (also when the non-RCT studies were excluded).187 Importantly, only one study reported long-term outcomes, which did not differ between groups.186
Endothelins, potent vasoconstrictors, have been implicated in CVS.49 ,50 ,51 An endothelin A receptor antagonist, clazosentan has been shown in experimental as well as clinical studies to ameliorate vasospasm.53 ,54 ,55 ,57 ,58 However, in a large multicentre, phase III study, no improvement in outcome could be shown.56 Similarly to nicardipine, patients receiving the drug suffered from a large number of systemic complications.
Local delivery
Administration of drugs targeting vasospasm is frequently hampered by systemic complications, a factor that has generated interest in local delivery methods. At present, the only locally delivered substance for which clinical data exist is nicardipine administered into the subarachnoid spaces as prolonged-release implants. Preliminary data comes from an open-label trial in Japan (n=97), where a decrease of the incidence of DCI was noted from 11% to 6%.188 Subsequently, in a single centre RCT (n=32), nicardipine implants were found to significantly decrease the incidence of vasospasm (73% vs 7%), delayed infarcts (47% vs 14%), as well as improved outcome (38% vs 6%).189 Notably, the study was conducted only on poor-grade SAH patients, hence, the generalisability and robustness of the results remains uncertain. Currently, no phase III study has confirmed the significance of the initial findings.
Prophylactic balloon angioplasty has not been shown in a multicentre phase II study to be beneficial following SAH.190 Therapeutic balloon angioplasty and intra-arterial vasodilators, while used in some patients when medical management has failed, are only now being studied in a randomised trial—Diagnostic and Therapeutic Management of Cerebral Vasospasm After Aneurysmatic Subarachnoid Haemorrhage (IMCVS) (clinicaltrials.gov; NCT01400360).
Drugs with multidirectional effects/neuroprotection
Statins, which have been shown to have diverse clinical effects, have been evaluated in four single-centre RCTs with mixed results.166 ,168 ,191–193 A meta-analysis of the trials has so far demonstrated no benefit from using this treatment.11 However, a multicentre study of simvastatin is currently ongoing (clinicaltrials.gov; NCT00731627), with another one comparing high dose vs low dose (clinicaltrials.gov; NCT01077206). Interestingly, statins have been shown to reduce the duration of impaired autoregulation after SAH, which has been implicated as a potential mechanism of action.166
Magnesium is another compound with neuroprotective effects which has been assessed in SAH.194 The interest in magnesium sparked from an observation that hypomagnesemia may be associated with increased incidence of DCI.195 However, the results from the largest multicentre study and a meta-analysis failed to demonstrate a significant difference in outcome.196
The neuroprotective effects of erythropoietin (EPO) have been studied in experimental models.197 ,198 Furthermore, EPO has been shown to ameliorate vasospasm and improve outcome after experimental SAH.199 Tseng,200 in a single-centre study demonstrated that, similar to statins, EPO treatment was associated with a reduced duration of impaired autoregulation, a lower incidence of severe vasospasm and DCI, as well as an improved outcome at discharge. However, long-term benefits were not demonstrated.
Albumin, 25%, has been shown to be neuroprotective.201 A pilot study of human albumin demonstrated a good tolerability. Results from phase III RCT are awaited.
Microthrombosis
The findings that SAH leads to clotting activation (physiological mechanism to prevent rebleeding), platelet aggregation and microthrombosis lead to design of studies of antiplatelet agents. Nevertheless, despite solid pathophysiological background, the results of the studies have been largely negative.151 Similarly, the role of low molecular weight heparin in the prevention of microthrombosis has been investigated in two RCTs. Results of the studies were mixed, with one demonstrating a lack of effect on outcome and four cases of intracranial bleeding thought to be related to the treatment.202 By contrast, another study found a beneficial effect of enoxaparin on vasospasm, DCI as well as outcome, with fewer haemorrhagic complication in the treated group.203 However, the results need to be treated with caution, as groups were not well matched for admission grade.
Free radicals and inflammation
Tirilizad mesylate, a non-glucocorticoid, 21-aminosteriod that inhibits lipid peroxidation, has been evaluated in four RCTs with mixed results.204–207 However, two meta-analyses demonstrated no effect on DCI, infarcts, or outcome.208 ,209
Three studies investigated the effect of free radical scavengers on DCI and outcome after SAH.210–212 Ebselen was found to improve outcome in a large study of 286 patients. Interestingly, the improvement in outcome was independent of the incidence of DCI which was unchanged between the treatment and placebo groups.211 Similarly, two other free-radical scavengers, nicaraven and edaravone, have been proven to be beneficial, after SAH.210 ,212
Several anti-inflammatory compounds have been studied after SAH. Suzuki,213 used a synthetic thromboxane synthetase inhibitor, OKY-046, to prevent DCI. They demonstrated reduction of DCI and improvement in outcome at 1 year.213 Methylprednisolone, a strong immunosuppressant, was shown in a randomised study to significantly improve outcome without any effect on vasospasm.214
Despite these promising results, none of the investigated compounds have been assessed in multicenter studies nor implemented in clinical practice.
Conclusions
Outcome form SAH has improved in the last 20 years. This is most likely due to early aneurysm repair, intensive management, and routine use of nimodipine. However, the exact influence of management of DCI on outcome is unclear. This is further complicated by the wide differences in the incidence of DCI, with some studies suggesting an incidence around 15–20%, while others as high as 40%. Despite new data and increased understating of the pathophysiology of SAH, DCI as well as EBI, no new treatments have been introduced since nimodipine. Data from large randomised controlled studies suggests that a pure focus on CVS, as the sole cause of DCI and poor outcome, is misguided. Nevertheless, the available data does not yet support other approaches aimed at mechanisms distinct form vasospasm, such as microthrombosis and platelet aggregation, inflammation and formation of free radicals. Consequently, current management strategies frequently focus on intensive care management with widespread use of pharmacological and interventional rescue therapies. While numerous targets are still being investigated, some of the more promising results come from drugs with multifactorial effects, such as statins or cilostazol. Overall, available data suggests that a focus on a single pathway may not be sufficient to improve outcomes in SAH. Furthermore, design of future clinical trials should take notice of the available findings and construct studies with appropriate selection of high-risk patients, as well as adequately sensitive and objective outcome measures. Similarly, studies which failed to demonstrate outcome benefits, where sound physiological data exist, should be re-evaluated with the aim of explaining the reason for futility, helping to define the patient groups which could benefit as well as provide background for future drug development.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online table 1
- Data supplement 2 - Online table 2
Footnotes
-
Contributors KPB: first draft, literature search, approved final version. MG: review of first draft and critical comments, approved final version. AH: review of first draft and critical comments, approved final version. TH: review of first draft and critical comments, approved final version. MC: review of first draft and critical comments, approved final version. RK: review of first draft and critical comments, approved final version. DKM: review of first draft and critical comments, approved final version. JDP: review of first draft and critical comments, approved final version, supervised project. PJK: review of first draft and critical comments, approved final version, supervised project
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.