Article Text

Abstract
Objective GNE myopathy is a rare recessive myopathy associated with inclusion bodies on muscle biopsy. The clinical phenotype is associated with distal muscle weakness with quadriceps sparing. Most of the current information on GNE myopathy has been obtained through studies of Jewish and Japanese patient cohorts carrying founder mutations in the GNE gene. However, little is known about GNE myopathy in Europe where the prevalence is thought to be very low.
Methods Patients were referred through the National Specialist Commissioning Team service for limb-girdle muscular dystrophies at Newcastle (UK). All patients harbouring mutations in the GNE gene were recruited for our study. Detailed clinical and genetic data as well as muscle MRIs and muscle biopsies were reviewed.
Results We identified 26 patients harbouring mutations in the GNE gene. Two previously reported mutations (c.1985C>T, p.Ala662Val and c.1225G>T, p.Asp409Tyr) were prevalent in the Scottish, Northern Irish and Northern English populations; with 90% of these patients carrying at least one of the two mutations. Clinically, we confirmed the homogenous pattern of selective quadriceps sparing but noted additional features like asymmetry of weakness at disease onset.
Conclusions GNE myopathy is an important diagnosis to consider in patients presenting with distal leg muscle weakness. We report, for the first time, two common mutations in the north of Britain and highlight the broader spectrum of clinical phenotypes. We also propose that the prevalence of GNE myopathy may be underestimated due to the frequent absence of rimmed vacuoles in the muscle biopsy.
- MUSCLE DISEASE
- MYOPATHY
- INCL BODY MYOSITIS
- NEUROMUSCULAR
- GENETICS
Statistics from Altmetric.com
Introduction
GNE myopathy is a rare inherited form of distal myopathy associated with inclusion bodies on muscle biopsy. In contrast with other forms of inclusion body myopathies,1–3 it is recessive in inheritance.4 ,5 The phenotypical descriptions of GNE myopathy include hereditary inclusion myopathy, quadriceps sparing myopathy, distal myopathy with rimmed vacuoles, and more rarely myopathy with facial diplegia or proximal weakness. Historical differences in nomenclature and classification may have led to an underestimation of the disease prevalence.6 Recently, the term glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase (GNE) myopathy has been coined to encompass all these phenotypical subgroups.
The diagnostic criteria of GNE myopathy include myopathy of early distal weakness, onset in early adulthood, mild or normal serum creatine kinase (CK) levels, rimmed vacuoles and detection of 14–18 nm filamentous inclusions.6 ,7 Inflammatory infiltrates are rarely seen in GNE myopathy in contrast with the sporadic form of inclusion body myositis.6 ,8 A definite diagnosis is conventionally confirmed by evidence of compound heterozygous or homozygous mutations in the GNE gene.
We report for the first time the clinical, histopathological and genetic findings of all the patients with genetically confirmed GNE myopathy, diagnosed through the National Specialist Commissioning Team (NSCT) service for limb-girdle muscular dystrophies at Newcastle.
Methods
Clinical assessment
A total of 26 patients were recruited via the Newcastle MRC Neuromuscular Centre, Institute of Genetic Medicine, Newcastle upon Tyne. Genetic testing for GNE myopathy in the UK is only available through Newcastle as part of the NSCT service. Some patients had testing on a research basis abroad before it became available as a diagnostic test in the UK; these patients have been retested by the NSCT service.
Comprehensive clinical histories and neurological examinations were completed during follow-up clinic visits. Newcastle institutional clinical research ethics board approval was obtained and all participants provided written informed consent for research.
Further investigations included serum CK levels, respiratory function (using spirometry), ECG and neurophysiological studies.
Muscle MRI was performed in seven patients. Axial and coronal planes of the pelvis and lower limbs were obtained using conventional T1-weighted spin echo sequences. Muscle biopsies were performed in 20 patients, 2 patients had repeat biopsies. Muscle histology was assessed by standard procedures including H&E staining and was processed for immunohistochemistry and multiplex Western blot.9
Sequence analysis
Genomic DNA was extracted from venous blood taken from patients and family members where available by automated DNA extraction on the M48 BioRobot using the MagAttract DNA blood Mini M48 kit (Qiagen 951336) as part of the routine service performed by the Northern Region Genetics service molecular laboratory.
Direct sequencing for coding exons 1–13, GenBank accession (NM_001128227.2) of the GNE gene was carried out plus more than 10 nucleotides of 5′ and 3′ intronic sequence. Alamut mutation interpretation software (Interactive Biosoftware, LLC, Rouen, France) was used to predict the pathogenicity of novel variants.
Results
Geographical and demographic characteristics
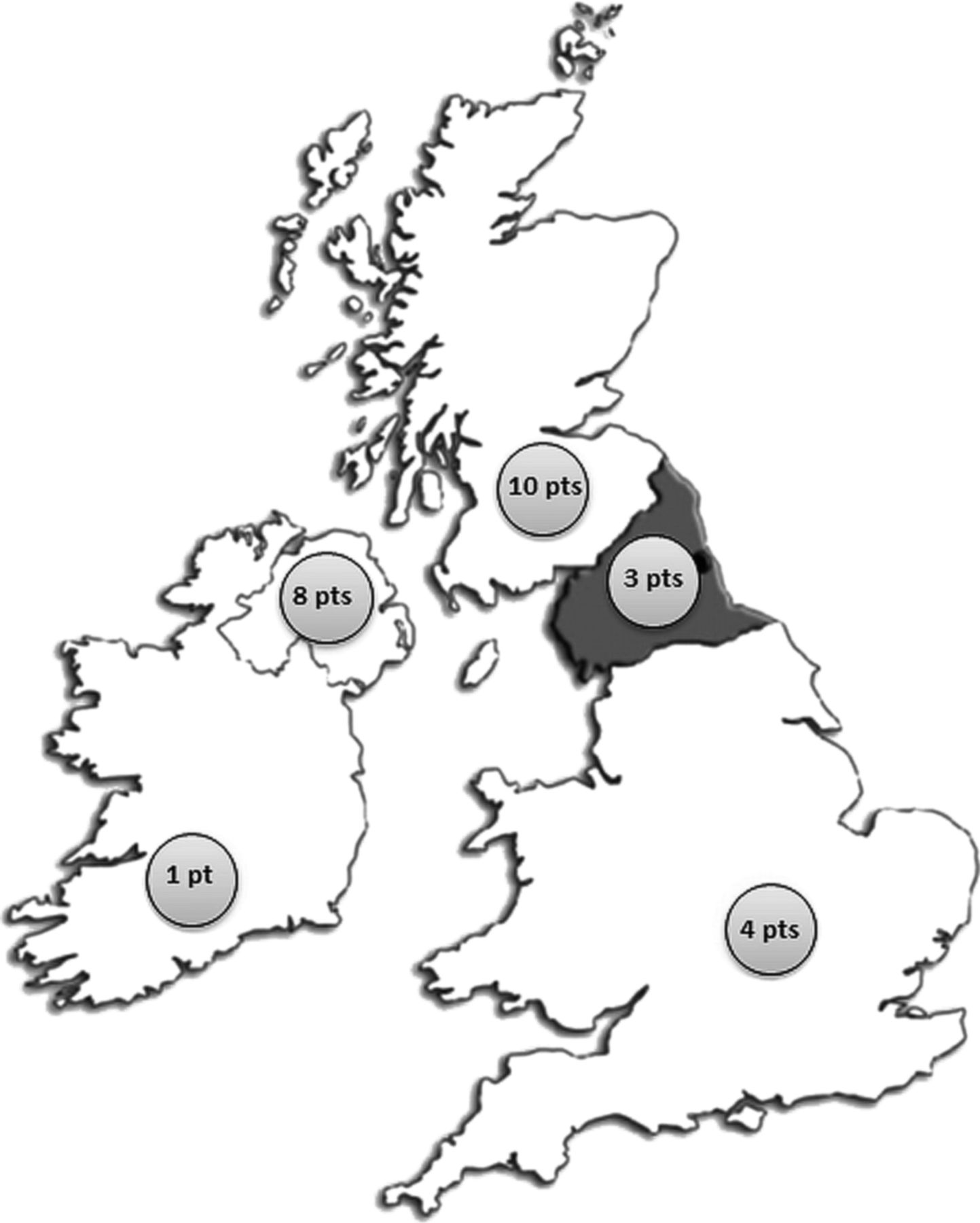
Genetic testing in Newcastle became available in September 2010 and a total of 87 full GNE gene screens have been performed since. Of the 87 referrals 64% were from England, 14% were from Northern Ireland and 13% were from Scotland. The remaining referrals were from abroad. In total, 26 patients with GNE myopathy were identified, 12 women and 14 men from 23 kinships referred between 1987 and 2013. Of the 26 patients 10 were from Scotland, 8 were from Northern Ireland and 7 from England. One patient was referred from the Republic of Ireland (figure 1).

Geographical distribution of patients with GNE myopathy referred to Newcastle between 1987 and 2012. A total of 87 full GNE gene screens have been performed in Newcastle since September 2010. Of the 87 referrals 64% were from England, 14% were from Northern Ireland and 13% were from Scotland. The remaining referrals were from abroad. The majority of patients with genetically confirmed GNE myopathy were referred from the North of Britain (Scotland, Northern Ireland and Northern England) with the highest numbers seen in Northern Ireland and Scotland.
Based on our data, the point prevalence of GNE myopathy in Britain (England, Scotland and Northern Ireland) is 0.04 per 100 000 population. The point prevalence for Scotland is 0.19 per 100 000 population, Northern Ireland 0.44 per 100 000 population and England 0.01 per 100 000 population. Of note the point prevalence of GNE myopathy in Japan is 0.3 per 100 000 population, one of the highest in the world. We accept that these figures need to be interpreted with caution in the absence of a more detailed population analysis and bearing in mind the challenges of disease ascertainment of GNE myopathy; however, it is worth noting that the estimated disease prevalence in Northern Ireland is close and possibly higher than in Japan.
Clinical features
The mean age of onset was 30±9.2 years (range 10–44 years). Fourteen patients presented in the fourth decade of life with two patients presenting in their fifth decade. The earliest age of onset reported was around the age of 10 years (table 1).
Clinical features and GNE genotypes of patients with genetically confirmed GNE-myopathy
Most patients presented with leg weakness (23/26). Three patients presented with calf muscle wasting, difficulties playing football and tripping, respectively. Most of those presenting with weakness had distal weakness (21/23). At the first clinical assessment 23/26 were found to have either unilateral (9/23) or bilateral (14/23) foot drop. It is worth noting that all patients with unilateral foot drop progressed to developing bilateral foot drop. Quadriceps sparing was evident at presentation in 21/26 patients and was still a feature at the most recent clinical assessment in 17 of these patients.
Scapular winging was observed in 11/26 patients being asymmetrical in 3 and symmetrical in 8 patients and was noted at onset of disease in 4 patients.
Most patients remained ambulant (21/26) with 5/26 being wheelchair dependent. The non-ambulant group was older with a mean age of 57.2±3.5 years. Ten patients with a disease onset of 15 years or more retained the ability to walk. One of these however needed to use a wheelchair outdoors. Involvement of upper limb muscles, proximal and distal, was seen in the majority of patients (22/26) on subsequent assessments (table 2).
Summary table of clinical features in patients with genetically confirmed GNE-myopathy
Serum CK was elevated in most patients (mean 447.9±321.8; range 75–1400 U/L) but was normal in five patients. Neurophysiological data was available for 14/26 patients. Myopathic features on electromyography (EMG) were seen in 11/14. Of the seven patients in whom we had detailed EMG reports, the myopathic features were most evident in the anterior compartment of the distal lower limb, correlating with the clinical pattern of distal muscle weakness. Neurogenic changes were reported in 3/14 patients.
Muscle pathology reports were available in 20 patients. All biopsies showed at least mild myopathic changes except in two, which were reported as normal or showed neurogenic features, respectively. Information on site of muscle sampling was available in eight patients with the femoral quadriceps muscle being the most common site. In three patients biopsies were sampled from the quadriceps and tibialis anterior muscles. Scattered cytoplasmic rimmed vacuoles/inclusions were identified in five patients only. There was evidence of inflammatory infiltrates in three biopsies. Muscle MRI of the lower limbs was carried out in seven patients (figure 2A–F). Most images exhibited varying degrees of symmetrical muscle atrophy with relative sparing of the quadriceps in all.
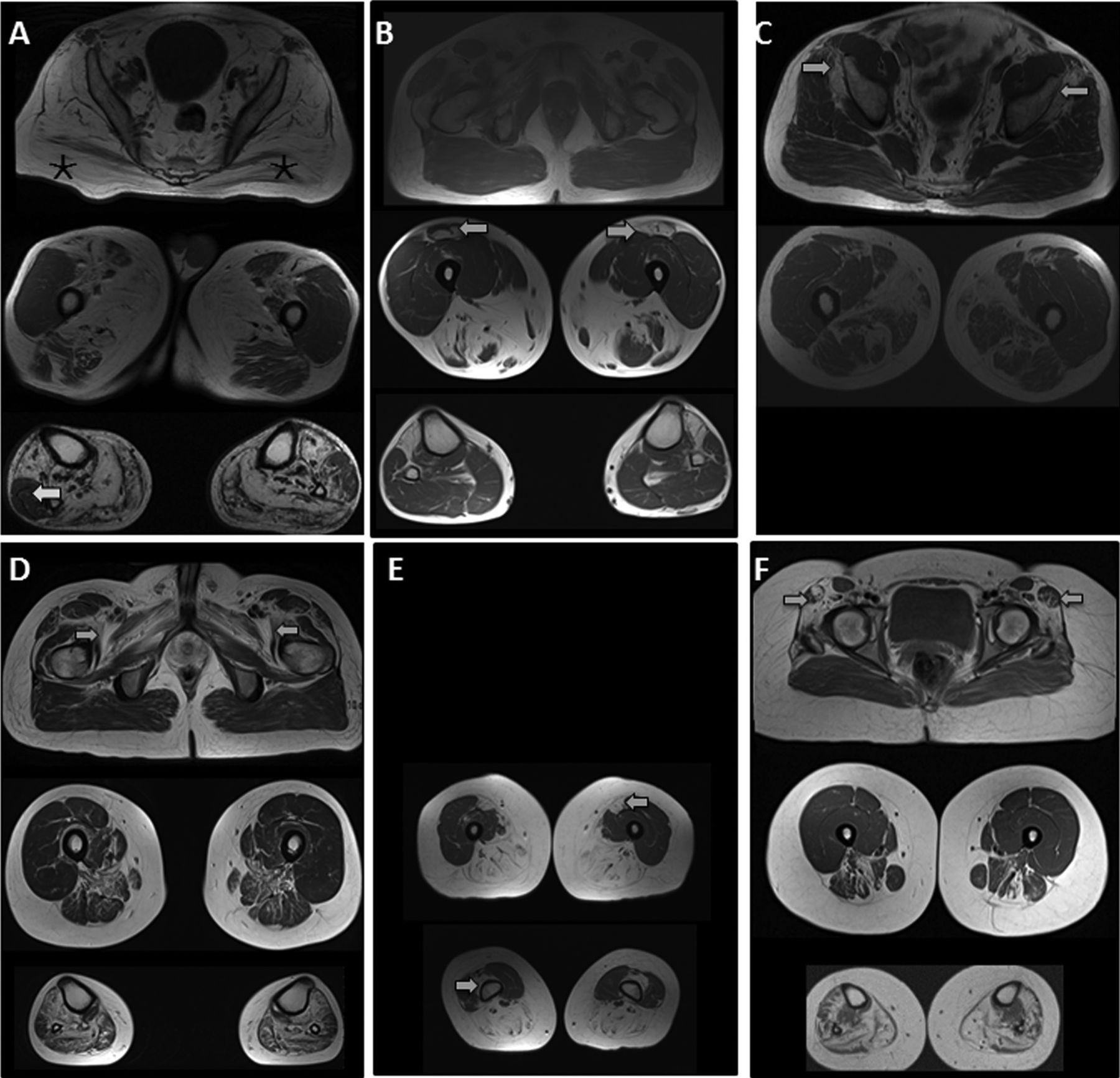
{kind=link}
{kind=link}
T1-weighted MR images of six patients with GNE myopathy. Evidence of quadriceps sparing in all patients with distal involvement of muscles in the anterior and posterior compartments. (A) Patient 15. Severe atrophy of both glutei maximi muscles (*). Left peroneus longus seems relatively spared in the left lower leg more than right (arrow). (B) Patient 10. Asymmetrical atrophy of rectus femoris muscles (arrows). (C) Patient 16. Early involvement of glutei minimi (arrows) with sparing of glutei maximi muscles. (D) Patient 19. Bilateral atrophy of iliopsoas, pectineus, adductor brevis and longus muscles (arrows). (E) Patient 9. Early atrophy of rectus femoris and vastus intermedius muscles (arrows). (F) Patient 5. Atrophy of tensor fascia lata muscles bilaterally (arrows).
Mutation analysis
Twenty-one patients were compound heterozygotes and five were homozygotes (in the absence of known consanguinity) with different GNE mutations (table 1). A total of 14 missense and 2 frameshift mutations were identified. Two frameshift mutations (c.805delG, Asp269Metfs*11 and c.1313dupT, p.Ser439Lysfs*6) and six missense mutations were novel (p.Ile618Thr, p.Gly393Glu, p.Asp111Tyr, p.Leu281Met, p.Gly86Arg, p.Val362Gly). Of note, a previous pathogenic mutation p.Val362Ala was reported at the same position as our mutation p.Val362Gly.10 Pathogenicity of the novel missense mutations was supported by parental testing showing segregation and/or in silico prediction tools (table 3). It is worth noting that two patients of Asian descent and a Northern English patient harboured the same heterozygous mutation (c.2179G>A, p.Val727Met).
Properties of GNE mutations identified in the British cohort
Two previously reported mutations (c.1985C>T, p.Ala662Val and c.1225G>T, p.Asp409Tyr) were highly prevalent in the Scottish, Northern Irish and Northern English populations. Both mutations were often found in the same patient in a compound heterozygous state. In five patients, mutation p.Ala662Val was found in a homozygous state. Notably, these two mutations were responsible for 33/52 pathogenic alleles (63%) in patients harbouring two mutations for the entire cohort, and 33/40 pathogenic alleles (83%) in patients from the north of the UK (Northern England, Scotland and Northern Ireland). Of patients from the north of the UK 90% (18/20) carried at least one of these mutations. We compared the clinical features of patients homozygous for p.Ala662Val (n=5) or compound heterozygous for p.Ala662Val and p.Asp409Tyr (n=10) with the remaining patients (n=11) and found no significant difference.
Discussion
GNE myopathy is an important diagnosis to consider in patients presenting with distal leg muscle weakness and elevated CK. This phenotype may be diagnostically challenging as it can mimic an inherited neuropathy or an alternative form of recessive distal myopathy like LGMD2B (Miyoshi phenotype), caused by dysferlin mutations.11 ,12 The sporadic form of inclusion body myositis shares important pathological features with GNE myopathy, namely the presence of inclusions on muscle biopsy, but should be reasonably distinguished from the inherited form, based on the later age of onset, the clear preservation of quadriceps muscles in GNE myopathy and evidence of inflammatory infiltrates in the majority of sporadic cases of inclusion body myositis.8 ,13
We describe a sizeable GNE myopathy cohort which exhibits similarities to other previously reported patients with regard to age of onset, quadriceps muscle sparing and the progressive nature of the disease.14–17 None of our patients developed respiratory insufficiency. The UK cohort is smaller than the previously described cohorts in Japan and Israel, and any conclusions are limited, but the clinical variability in age of onset and progression appears to be greater in the UK. We also note some asymmetry of weakness (unilateral food drop and scapular winging) at onset in several patients and slow disease progression. More than half of our patients with a disease duration of more than 15 years remained ambulant. Quadriceps sparing and asymmetry of muscle involvement were noted on muscle MRI and correlated with clinical signs.
In our cohort, the mean delay in diagnosis from symptom onset was 17.21±11.9 years (range 3–53 years). With the introduction of genetic testing in the UK in 2010, molecular confirmation became more achievable leading to shorter diagnostic delays.
Although rare, inflammatory markers on muscle biopsy may be present in GNE myopathy, predominantly at early stages, and cannot therefore exclude a genetic cause of the muscle pathology.18
Previous studies have indicated a possible association of homozygous kinase domain mutations in the context of early disease onset, and low level CK with a higher risk of respiratory failure.19 None of our patients developed respiratory insufficiency. One patient was reported to have mild systolic dysfunction and further investigations are awaited to ascertain its significance. Interestingly, the Northern Irish patients harbouring the p.Ala662Val mutation exhibited a particular gradient of weakness in the distal deep finger flexors with more involvement of the index finger compared with the little finger in both hands. To our knowledge, this is the first description of such selective muscle involvement in the distal upper limbs and seems to correlate with the genotype of the patient.
More than 60 mutations in the GNE gene have been associated with muscle pathology.4 ,20–22 In our cohort, mutation analysis revealed a number of novel missense (p.Ile618Thr, p.Gly393Glu, p.Asp111Tyr, p.Leu281Met, p.Gly86Arg and p.Val362Gly) and frameshift variants (c.805delG, Asp269Metfs*11 and c.1313dupT, p.Ser439Lysfs*6) spanning the length of the GNE gene with no suggestion of hot spots nor mutations in the allosteric domain.4 ,10 ,22–24
We have not identified any of the common mutations previously described in the Jewish and Japanese populations. Interestingly, we identified two very common mutations (c.1985C>T, p.Ala662Val and c.1225G>T, p.Asp409Tyr) in 18 out of the 20 patients diagnosed in Northern England, Scotland and Northern Ireland; with 5 homozygotes carrying the c.1985C>T, p.Ala662Val, 10 patients were compound heterozygotes for both mutations. The c.1225G>T, p.Asp409Tyr mutation had previously been reported in five individuals: two from the USA, two from Scotland and one from Ireland.10 ,22 This is in keeping with the hypothesis that p.Asp409Tyr is a Northern British founder mutation. The c.1985C>T, p.Ala662Val mutation has previously been described in other populations of German, Irish, Jewish, American, Scottish, Japanese and more recently Chinese background.4 ,10 ,16 ,22 ,24–26 Therefore, this mutation may be generally very frequent or may have arisen several times in different populations. It is also worth noting that population migration and immigration in Northern Ireland and Scotland tend to be less than in England, potentially affecting the gene pool. The predominance of these two mutations in the Northern British population is very striking and one should clearly consider a diagnostic strategy which may include early testing for the common GNE mutations in the UK and probably other parts of Europe.
GNE encodes for a bifunctional enzyme (uridine diphospho-N-acetylglucosamine 2-epimerase/N-acetyl-mannosamine kinase (GNE/N-acetylmannosamine kinase (MNK))) that catalyses the first two steps of sialic acid biosynthesis.5 ,7 Sialic acid is the most abundant terminal monosaccharide on glycoconjugates in eukaryotes.27
Complete loss of GNE expression is probably incompatible with life as evidenced by embryonic death seen with knockout inactivation of GNE in mice. In keeping with this hypothesis, all patients with GNE myopathy identified so far carried a missense mutation at least on one of the mutated alleles, allowing for residual enzyme activity.
Remarkably in our cohort, two mutations seem to be very common: p.Ala662Val located in the kinase domain and p.Asp409Tyr positioned in the epimerase domain. Both these mutations have previously been reported and functionally characterised in vitro.28 ,29 Mutation p.Asp409Tyr was shown to result in a 60% reduction of epimerase activity and an 80% reduction of kinase activity while p.Ala662Val mutation displayed only slight reduction in kinase and epimerase activities. In our patient cohort, patients homozygous for the Ala662Val had ‘milder’ features with most presenting with unilateral foot drop and four out of five remaining ambulant after 3–19 years from disease onset. While patients carrying one heterozygous p.Asp409Tyr mutation were more likely to present with bilateral foot drop (7/10) and become non-ambulant with 3 out of the 5 non-ambulant patients in our cohort harbouring this heterozygous mutation.
Finally, it is essential that accurate clinical and genetic data continue to be collected on patients with GNE myopathy across the world to allow effective assessment of any new emerging therapeutic agents.
Web-based databases (accessed June 2013): http://www.ons.gov.uk, http://www.gro-scotland.gov.uk, http://www.nisra.gov.uk, (2011 census), http://www.interactive-biosoftware.com/, http://www.dmd.nl/nmdb2/home.php?select_db=GNE, http://www.clinicaltrials.gov/ct2/show/NCT01634750?term=GNE+myopathy&rank=2. http://www.treat-nmd.eu/gne/patient-registries/patient-registries/ and http://www.treat-nmd.eu/
Acknowledgments
The authors thank the patients and clinicians who took part in this study, in particular Dr Michela Guglieri for her input in collecting some of the clinical data in this study. Diagnostic facilities at the Newcastle Muscle Centre are supported by the National Specialised Commissioning Team (NSCT) for rare neuromuscular disorders. The Institute of Genetic Medicine in Newcastle is part of the MRC Centre for Neuromuscular Diseases and the TREAT-NMD Alliance (http://www.treat-nmd.eu).
References
Footnotes
-
AC and KMB contributed equally to this study.
-
Contributors AC and KMB: Design and conceptualisation of study, data acquisition, interpretation and analysis (molecular and clinical data), manuscript authorship. JH: Data acquisition, interpretation and analysis (molecular data), revising of manuscript critically for important intellectual content. CL, JM, PM, RP, WS, JW, AS, TE, OP, ME, VS and KB: Data interpretation and analysis (clinical data), revising of manuscript critically for important intellectual content. TP and EH: data interpretation and analysis (histopathological data) and revising of manuscript critically for important intellectual content. HL: Design and conceptualisation of study, data acquisition, interpretation and analysis (molecular and clinical data), manuscript authorship and revising for important intellectual content. All coauthors have provided their final approval of the version to be published.
-
Competing interests None.
-
Ethics approval Newcastle institutional clinical research ethics board.
-
Provenance and peer review Not commissioned; externally peer reviewed.