Article Text

Abstract
Peripheral nerve diseases are traditionally classified as demyelinating or axonal. It has been recently proposed that microstructural changes restricted to the nodal/paranodal region may be the key to understanding the pathophysiology of antiganglioside antibody mediated neuropathies. We reviewed neuropathies with different aetiologies (dysimmune, inflammatory, ischaemic, nutritional, toxic) in which evidence from nerve conductions, excitability studies, pathology and animal models, indicate the involvement of the nodal region in the pathogenesis. For these neuropathies, the classification in demyelinating and axonal is inadequate or even misleading, we therefore propose a new category of nodopathy that has the following features: (1) it is characterised by a pathophysiological continuum from transitory nerve conduction block to axonal degeneration; (2) the conduction block may be due to paranodal myelin detachment, node lengthening, dysfunction or disruption of Na+ channels, altered homeostasis of water and ions, or abnormal polarisation of the axolemma; (3) the conduction block may be promptly reversible without development of excessive temporal dispersion; (4) axonal degeneration, depending on the specific disorder and its severity, eventually follows the conduction block. The term nodopathy focuses to the site of primary nerve injury, avoids confusion with segmental demyelinating neuropathies and circumvents the apparent paradox that something axonal may be reversible and have a good prognosis.
- NEUROPATHY
- NEUROPHYSIOLOGY
- GUILLAIN-BARRE SYNDROME
Statistics from Altmetric.com
Introduction
In 1871, Louis Antoine Ranvier described the ‘étranglements annulaires’ of nerve fibres, which were soon called the ‘nodes de Ranvier’.1 He suggested that the nodes were involved in the exchange of nutrients between blood and nerve fibres. It took almost eight decades to demonstrate that the nodes of Ranvier are, in myelinated fibres, the sites where inward membrane currents are generated to sustain saltatory conduction.2 The node of Ranvier is currently regarded as one of the crucial structures for functioning of the mammalian nervous system, ensuring the rapid and long distance transmission of impulses with the least expenditure of energy.
The diseases of peripheral nerves are traditionally classified as demyelinating or axonal. It has been recently proposed that dysfunction and disruption of the nodal region may be the key to better understanding of the pathophysiology of antiganglioside antibody-mediated neuropathies and other neurological disorders.3 ,4 In this review, after summarising the basic concepts on the architecture, molecular organisation and biophysical properties of the nodal region, we report disorders with different aetiology but sharing the involvement of the nodal region in the pathogenesis.
Anatomy, biophysical properties and molecular organisation of the nodal region
The myelinated fibres are organised in distinct domains: node, paranode, juxta-paranode and inter-node. At the node (about 1 μm in length), the myelin is interrupted and the axolemma is in direct contact with the extracellular fluid, though still covered by Schwann cell microvilli. At the paranode, the uncompacted myelin loops are tightly attached to the axolemma and at internodes (1–2 mm in length) the axons are surrounded by compact myelin (figure 1A).
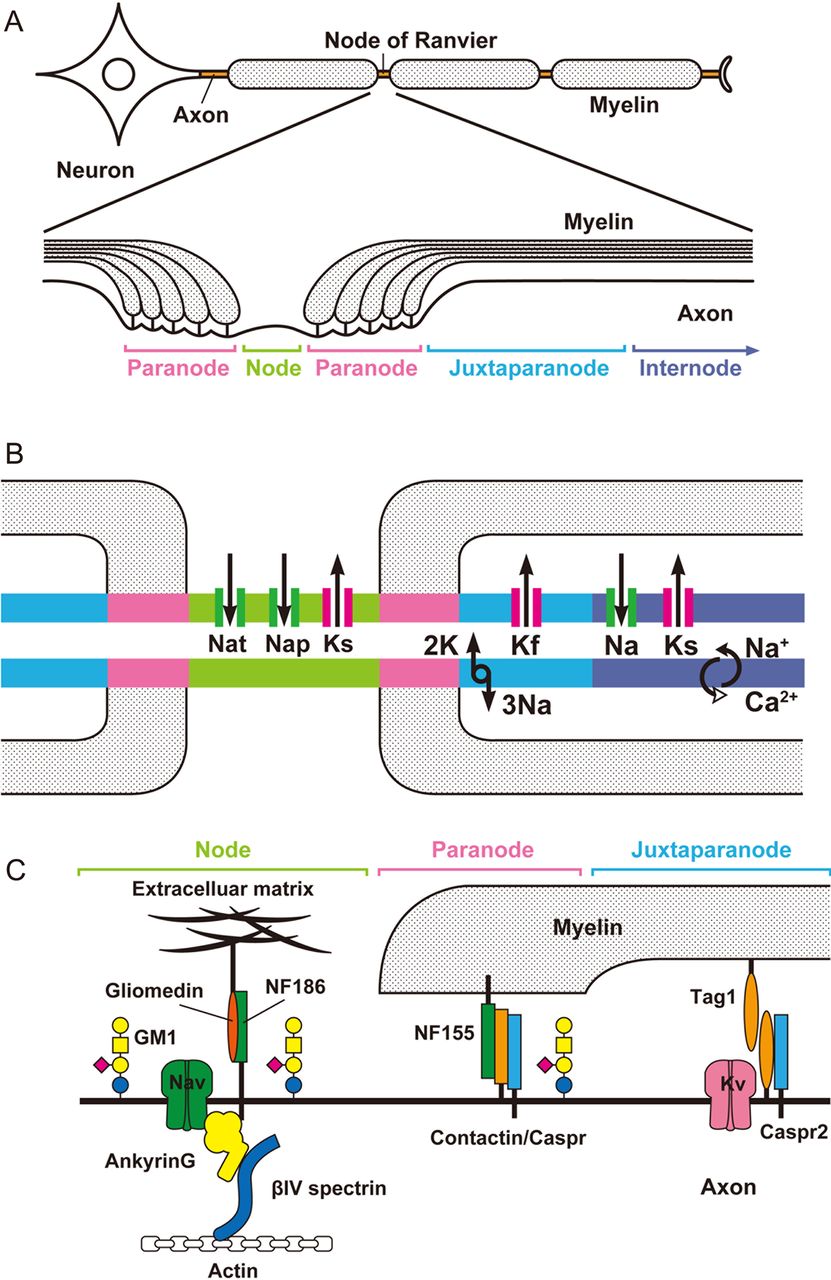
(A) Anatomical organisation of myelinated nerve fibre and its subdomains. (B) Schematic representation of components affecting excitability of axolemma. Distribution of voltage-gated-ion-channels and their current directions: Nat, transient Na+ current; Nap, persistent Na+ current; Ks, slow K+ current; Kf, fast K+ current. The Na+/K+ pump removes 3 Na+ from the axoplasma in exchange for 2 K+. The Na+/Ca2+ion exchanger removes Ca2+ from the axoplasm but can reverse its action. (C) Molecular organisation at nodes, paranodes and juxtaparanodes. NF, neurofascin, Caspr, Contactin associated protein. TAG-1, transient axonal glycoprotein 1. GM1 ganglioside is enriched at nodal and paranodal axolemma.
The axolemma of mammalian myelinated axons has a highly ordered molecular structure with a non-uniform distribution of ion channels (figure 1B). The node has a high density of voltage-gated Na+ channels (1000–2000/μm2) and slow K+ channels.5 The juxta-paranode contains a high density of voltage-gated fast K+ channels. The internode has the largest absolute number of Na+ channels, and fast and slow K+ channels, although their density is relatively low. For the Na+ channel the density is <25/μm2, which is not sufficient to sustain the propagation of the action potential. Na+ channels are of the Nav1.6 type and contribute to transient and persistent currents. A number of subtypes of Na+ channels are expressed in the peripheral nervous system, but only Nav1.6 is found at the nodes of Ranvier in adult mammalian myelinated axons.6 Whereas K+ channels are the largest and most diverse group of ion channels, represented by over 70 known loci in the mammalian genome.7 physiologically, axonal K+ channels consist of five types, sustaining fast, intermediate and slow currents with overlapping properties.8 In human axons, the resting membrane potential is −75 to −80 mV and determined by the equilibrium potential for K+ ions and to a lesser extent by the equilibrium for Na+ ions and the Na+/K+ pump. The term depolarisation indicates a shift in membrane potential toward more positive values than the resting membrane potential, whereas hyperpolarisation indicates a shift toward more negative values. Transient Na+ channels generate 98–99% of the total nodal inward ionic current that depolarises the membrane potential and generate the action potential. Persistent Na+ channels remain open over a wider range of membrane potentials and determine threshold for excitation. Fast K+ channels open 0.5–1.0 ms later than the transient Na+ channel, passing an outward K+ current, which takes back the membrane potential towards the resting value or hyperpolarises it. Slow K+ channels are concentrated at the node and open slowly, in the order of tens of ms, after depolarisation, modulate repetitive firing and induce repolarisation in response to prolonged depolarisation. The Na+/K+ pump restores transmembrane gradients of Na+ and K+, it is electrogenic and contributes to make the membrane potential more negative because at each cycle it expels three Na+ ions for every two K+ ions brought into the axon.5 The pump requires energy and hydrolises one ATP molecule with each cycle; its localisation remains controversial. The Na+/Ca2+ exchanger extrudes Ca2+ from the axoplasm in exchange for Na+ maintaining Ca2+ homeostasis.5 When the Na+ gradient across the membrane is reversed, the exchanger can run in the reverse mode, leading to increased levels of intracellular Ca2+.9
Figure 1C is a simplified representation of the molecular organisation at the nodal region. The nodal Na+ channels are anchored to spectrin of the axonal cytoskeleton via ankyrinG and to gliomedin of the Schwann cell microvilli via neurofascin-186.10 At the paranode, axonal contactin-associated protein (Caspr) and contactin are tightly connected to neurofascin-155 of paranodal myelin loops. This complex forms septate-like junctions, which separate the nodal Na+ channel clusters from the juxtaparanodal K+ channels. The juxtaparanodal K+ channels are anchored by Caspr-2 and transient axonal glycoprotein 1 (TAG-1), which is an adhesion molecule that is expressed both on axolemma and opposing Schwann cell membrane. The interaction between TAG-1 on the axolemma with Caspr and TAG-1 on the Schwann cell membrane is crucial for clustering of juxtaparanodal Kv1.1 and 1.2 channels. Gangliosides are glycolipids consisting of a lipid ceramide component located in the bilipid membrane and an extracellular sugar portion to which 0, 1, or more sialic acid residues may be attached. High-resolution light and electron microscopy of teased fibres from rat spinal roots and sciatic nerves has shown gangliosides on the following structures: GM1 on nodal and paranodal axolemma, abaxonal Schwann cell membrane, and Schwann cell microvilli and GD1a on nodes and abaxonal Schwann cell membrane.11 Gangliosides probably interact with the nodal proteins and contribute to the stability of axon-glia interface at the paranode, as in GM1 and GD1a knockout mice the paranodal myelin loops do not attach to the axolemma, Na+ channel cluster are disrupted, and K+ channels are mislocated to the paranode.12
The node and saltatory conduction
The action potential is generated at the axon initial segment, which shares a common molecular organisation with the nodes of Ranvier, and propagates jumping from one node to the following one (figure 2A–C). The safety factor for impulse transmission is the ratio of the driving current to threshold (the required current to depolarise the membrane and induce an action current). In normal myelinated axons this factor is 5–10 and to generate an action potential has to be more than one. Computer simulations showed that conduction is still possible if myelin thickness is 2.7% of normal at one internode or 4% at two adjacent internodes.13 In figure 2D, the effects of segmental demyelination are schematised up to conduction failure. In any case, studies of experimentally induced focal demyelination showed that minimal involvement of paranodal myelin suffices to block nerve conduction, emphasising the crucial role of the paranode in saltatory conduction.14 Restored conduction after demyelination may be continuous by increased expression and density of Na+ channels on the denuded internode, microsaltatory with formation of new Na+ channel cluster and saltatory, although slow, due to remyelination with the development of short internodes.15–17

Saltatory conduction in normally myelinated and demyelinated axons. (A) At the active node (left), transient Na+ channels are open, inducing an inward ionic named action current. This causes a current circuit that leads to an outward capacitive ionic driving current at the successive node to be activated (right). (B) Positive charges accumulate at the inside of the node to be activated and are withdrawn at its outside; this depolarises the membrane. (C) As soon as the node membrane is depolarised to threshold, its Na+ channels open, Na+ enters into the axon and induces further depolarisation in a regenerative process. The driving current converts into an action current generating an action potential. (D) If the myelin sheath is damaged the driving current leaks through the damaged myelin sheath (arrows) and dissipates over a large area including the paranode and the juxtaparanode, with increased capacitance impairing the depolarisation of the node to be activated. Depolarisation is additionally impaired because the driving current activates the exposed juxtaparanodal fast K+ channels (K), shifting the membrane potential toward more negative values, preventing the reaching of the threshold and shortening the duration of the action potential. If the safety factor is below 1 there is block of impulse transmission. Plus sign (+) represents positive charges. Modified from Franssen and Straver 2013.73
Focusing on the nodal region, the possible alterations that may reduce the safety factor of transmission and block nerve conduction are schematised in figure 3.
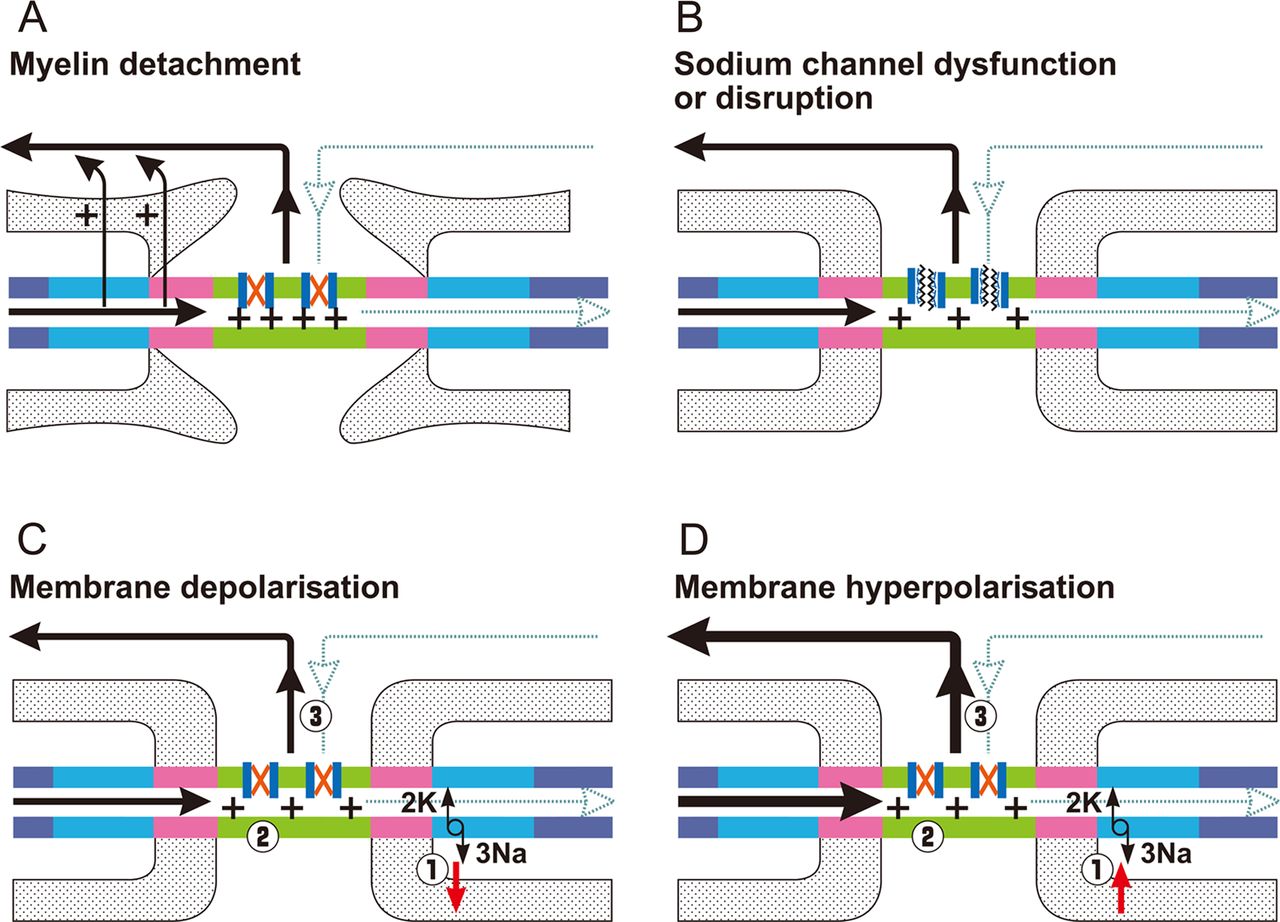
Nodal causes of block of impulse transmission. The black line is the driving current and the dotted blue line is the action current that is impaired. The red X within the Na+ channel indicates that the channel remains closed. (A) Detachment of the terminal myelin loops causes current leakage (arrows) and dissipates the driving current over an increased area with larger capacitance. Consequently, depolarisation may not suffice to activate the required number of Na+ channels and the action current is not generated. (B) Dysfunction/disruption of sodium channel (depicted with a deranged structure) directly impairs the generation of the action current. (C) Reduced functioning of Na+/K+ pumps (1) induces persistent depolarisation of the membrane potential with inactivation of transient Na+ channels (2) and impairment of action current (3). (D) Increased functioning of Na+/K+ pumps (1) induces persistent hyperpolarisation of the membrane potential (2) and more driving current (depicted with a thicker line) is needed (3) to reach threshold current to open Na+ channels. Focal alterations of resting membrane potential as in C and D may coexist at contiguous nerve segments. Modified from Franssen and Straver 2013.73
Nodal involvement and axonal degeneration
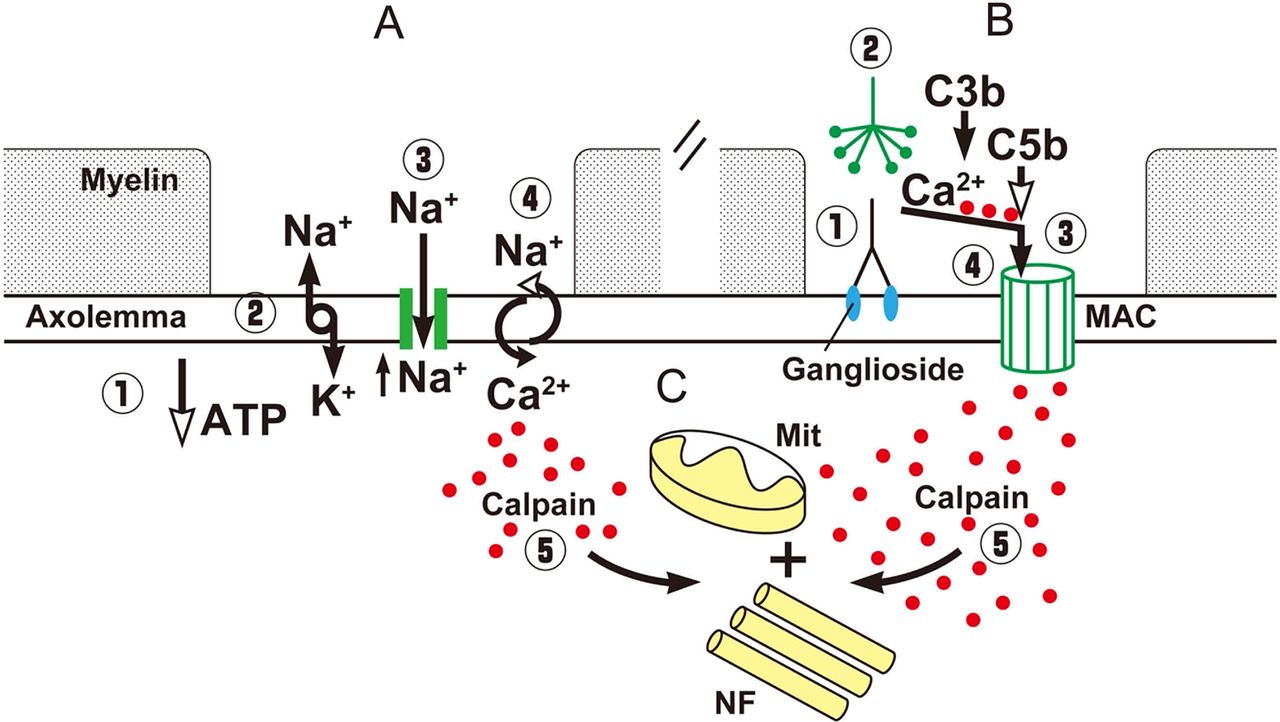
Alterations at the nodal region may induce axonal degeneration. For example, malfunctioning of the Na+/K+ pump due to ATP depletion by various causes results in axoplasmic Na+ accumulation and membrane depolarisation, which activates nodal persistent Na+ channels inducing further depolarisation. This, in turn, increases the percentage of inactivated transient Na+ channels and induces failure of conduction (figure 3C). If this process persists, Na+ accumulation reverses the axolemmal Na+/Ca2+ exchanger to remove the excess Na+ in exchange for Ca2+, which enters into the axon (figure 4A).7 Moreover, intense Na+ influx activates voltage-dependent Ca2+ influx and stimulates mitochondria to release additional Ca2+.18 The resulting axoplasmic Ca2+ accumulation may activate calpain, a ubiquitous calcium sensitive protease, causing proteolytic cleavage of neurofilaments, mitochondrial damage and, finally, Wallerian degeneration (figure 4A).19 ,20
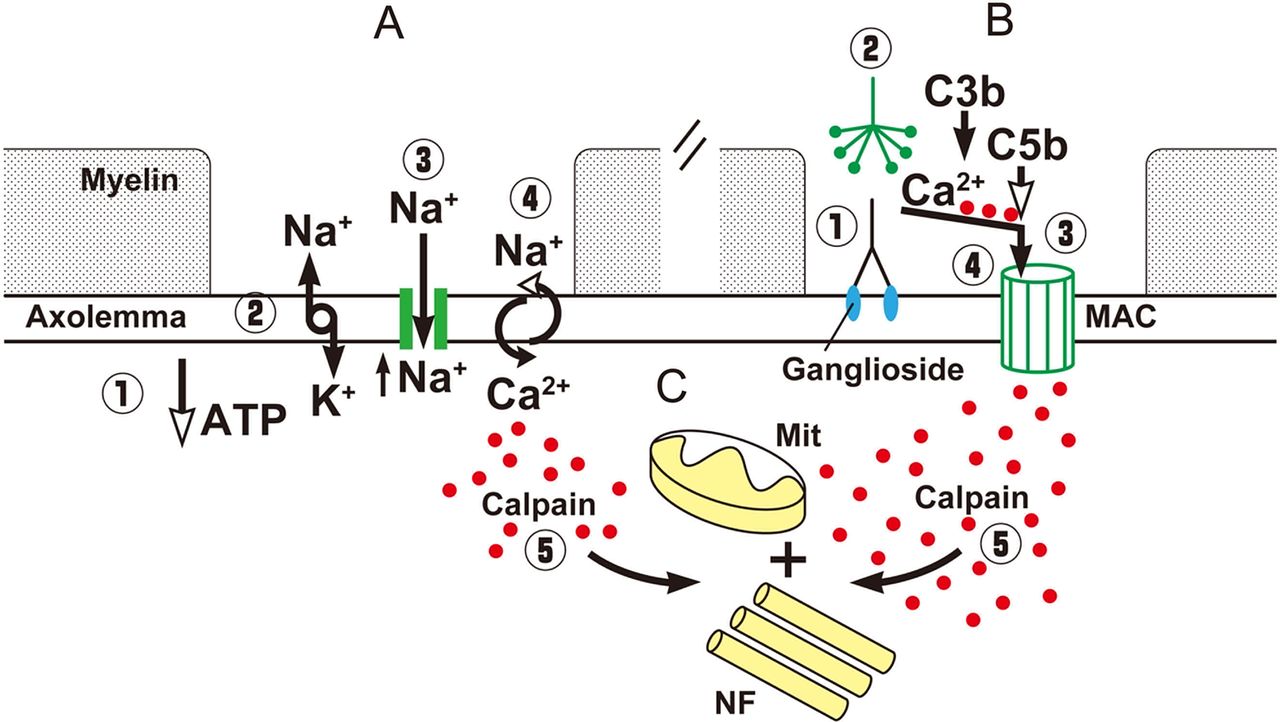
Schematic diagram of the possible events leading to axonal degeneration in nodoparanodopathies. (A) Energy failure, induces depletion of ATP (1), this reduces the function of the NA+/K+ pump with increase of axoplasmic NA+ and membrane depolarisation (2), which activates persistent Na+ channels with further NA+ influx and accumulation (3), activity of Ca2+/Na+ exchanger is reversed and Ca2+ (red dots) accumulates in the axoplasma (4). (B) Antibodies bind to gangliosides in the axolemma (1), the classical pathway of complement is activated (2), the terminal components of complement form the membrane attack complex (MAC) pore (3), Ca2+ enters through the pores and accumulates in the axoplasma. (C) Common final pathway of axonal degeneration with activation of Ca2+-dependent calpain (5) causing proteolytic cleavage of neurofilaments (NF), damage of mitochondria (Mit) and, finally, Wallerian degeneration.
Nerve conduction and excitability studies in disorders involving the nodal region
For more than 50 years, motor and sensory conduction studies have been used for investigating peripheral nerve function. Routine studies employ supramaximal stimulation to measure latency and amplitude of compound muscle action potential (CMAP) and sensory nerve action potential (SNAP), and to calculate conduction velocity of the fastest fibres. Some common beliefs such as that conduction block (CB) and conduction velocity slowing are present only in demyelinating neuropathies may be misleading in the interpretation of the electrophysiological results in disorders affecting the nodal region. CB is commonly defined as the failure of a nerve impulse to propagate through a structurally intact axon, and practically demonstrated by comparing amplitudes and areas of CMAPs evoked by two sites of stimulation. CB is considered one of the electrophysiological correlates of demyelination (figure 5A), however, conditions affecting the excitable axolemma at the nodal region may also induce CB (figures 3 and 5B). In this case, the arrest of nerve conduction has been called axonal CB.21 Increased duration and fragmentation of CMAP, called temporal dispersion (TD), help to distinguish demyelinating and axonal CB (figure 5A, B). Remyelination after acute demyelination, or ongoing demyelination and remyelination, are characterised by desynchronisation of conduction among fibres, which induces excessive TD, and decreased amplitude and area of CMAP, because of cancellation between opposing phases of single motor unit potentials (figure 5A). On the other hand, axonal CB may promptly reverse without TD of CMAP (figure 5B, D).22 ,23 Slow conduction velocity is thought to be characteristic of a demyelinating neuropathy and in axonal neuropathies conduction velocity is usually considered to be normal or slightly slow. However, partial inactivation of the Na+ channel, by intravenous infusion of lidocaine in humans, reduces the conduction velocity of single motor unit potential to 20–30 m/s, possibly by increasing the rise time of the action potential and the internodal conduction time.24 This explains why conduction velocity may be slow in disorders affecting the node and why conduction velocity improves in parallel with the resolution of CB (figure 5B). Reduced amplitude of distal CMAP, in the absence of demyelinating features, is usually considered indicative of axonal degeneration (figure 5C) but may also be due to CB in terminal axons where the safety factor is physiologically reduced due to distal branching (figure 5D). Looking at figure 5, it is striking that a correct distinction between a demyelinating and axonal CB or between axonal degeneration and CB in terminal axons can be made only after serial recordings, which are not usually carried out in everyday practice.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
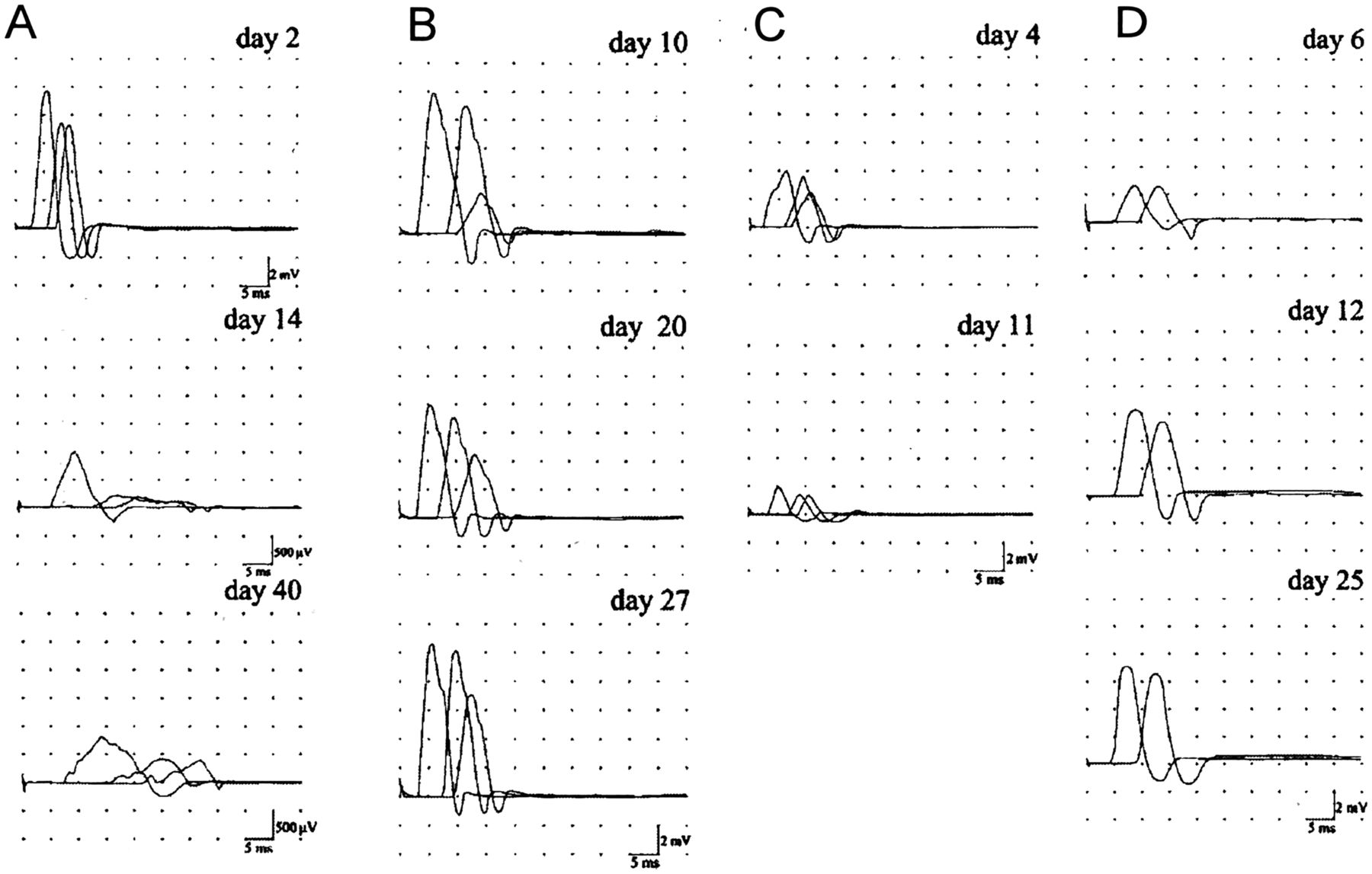
Superimposed compound muscle action potentials (CMAPs) recorded from the abductor digiti minimi after ulnar nerve stimulation at the wrist, below-elbow and above-elbow and from the abductor pollicis brevis after median nerve stimulation at the wrist and elbow. (A) Acute inflammatory demyelinating neuropathy: ulnar nerve. On day 2, all conduction parameters were normal. On day 14, all CMAPs were dispersed, distal CMAP amplitude was greatly reduced (1 mV), distal motor latency was increased (5.7 ms), the CMAP amplitude ratio between below-elbow and wrist stimulation was 0.2, and conduction velocities were reduced (20 m/s in the below-elbow wrist segment and 26 m/s across the elbow). On day 40, the CMAP amplitude ratio between below-elbow and wrist stimulation was 0.5 but all CMAPs were still reduced in amplitude and dispersed, DML was further increased (7.2 ms) and conduction velocities reduced (19 m/s in the below-elbow wrist segment and 16 m/s across the elbow). (B) Acute motor axonal neuropathy with conduction block (CB): ulnar nerve. On day 10, the CMAP amplitude ratio across the elbow (0.3) indicates CB. The ratio improved in the following recording up to 0.7 on day 27 without the development of excessive temporal dispersion. Conduction velocity across the elbow was slow (38 m/s) on day 10 but improved in parallel with the resolution of CB (48 m/s on day 27). (C) Acute motor axonal neuropathy with axonal degeneration: ulnar nerve distal CMAP amplitude was already decreased (4 mV) on day 4 and further decreased (2 mV) on day 11 to remain stable. (D) Acute motor axonal neuropathy with distal CB: median nerve. On day 6, distal and proximal CMAP amplitudes were reduced (2.6 mV). On day 12, distal CMAP was increased by 142%, returning to within the normal range. There was no excessive temporal dispersion of proximal or distal CMAP in all recordings. Modified from Uncini et al.34
Studies of axonal excitability, not yet widely employed, have the potential to clarify the biophysical abnormalities underlying the defects in impulse conduction. In the 1990s, a threshold tracking technique was developed to non-invasively measure several indices of axonal excitability, such as refractoriness, supernormality, late subnormality, threshold electrotonus and strength-duration properties.25 These indices depend on the biophysical properties of the axonal membrane at the site of stimulation and can provide an indirect insight into resting membrane potential and Na+ or K+ channel function. By delivering a conditioning stimulus, which alters membrane potential or activates specific ion channels, the current required to produce a threshold potential will change. Automated computerised tracking of a ‘threshold’ for multiple nerve excitability measurements is currently available and allows testing of a motor or sensory nerve within 10 min. The major limitations of excitability testing methods are that: (1) only sites where the peripheral nerve is superficial can be evaluated, (2) excitability properties can be assessed only at the site of stimulation, and (3) nerve conduction between the stimulus and recording sites should be secure; if CB develops, nerve excitability testing no longer works. In spite of limitations, axonal excitability studies have proved to be useful in the understanding of the pathophysiology of disorders affecting the nodal region.
Nodopathies of the peripheral nerve
The term nodo-paranodopathy was originally proposed to better characterise neuropathies with antiganglioside antibodies characterised by a common pathogenic mechanism of dysfunction/disruption at the node of Ranvier resulting in a pathophysiological continuum from transitory nerve conduction failure to axonal degeneration.3 We here extend this concept to include neuropathies of different aetiology (dysimmune, inflammatory, ischaemic, nutritional and toxic) in which the involvement of the nodal region is determinant. As the pathogenic mechanisms are mainly focused on the node, we propose to classify these neuropathies as nodopathies.
Acute motor axonal neuropathy
Acute motor axonal neuropathy (AMAN), the primarily axonal Guillain-Barré syndrome (GBS) subtype, is associated with a preceding Campylobacter jejuni infection and IgG antibodies against GM1 in 64% of patients, GM1b in 66%, GD1a in 45% and GalNac-GD1a in 33%.26 More recently, antibodies directed exclusively against complex gangliosides such as GM1/GalNAc-GD1a and LM1/GA1 have been described.27 AMAN was initially characterised by simple axonal degeneration and its electrodiagnosis was based on reduced distal CMAP amplitudes and absence of demyelinating features (figure 5C).21 However, patients with anti-GM1 antibodies showing CB/slowing that promptly resolved without the development of excessive TD of CMAPs were reported (figure 5B, D).22 ,23 Owing to rapid recovery and lack of slow components characteristic of de-remyelination, this CB was thought to be caused by an impaired conduction at the nodes of Ranvier, possibly due to temporary loss of Na+ channel function, and named reversible conduction failure or axonal CB to distinguish it from demyelinating CB.21 ,22 Excitability studies of the median nerve at the wrist showed increased refractoriness that rapidly normalised in parallel with a recovery of CMAP amplitude suggesting conduction failure of the second impulse distal to the wrist, possibly in distal axonal branches or axonal terminals.28
The finding of nodal IgG deposition, complement and membrane attack complex (MAC) formation in humans and AMAN rabbits indicated a direct injury of the nodal axolemma with subsequent axonal degeneration.29–31 Paranodal myelin detachment and nodal lengthening mimic paranodal demyelination but the injury site is primarily axonal and nodal. MAC forms pores in the axolemma that allow massive, not selective, influx of water and ions into the axon. Ex vivo incubation of mouse distal motor axon with anti-GD1a antibodies and complement resulted in complement deposits at the nodes, MAC formation, loss of nodal Nav1.6, ankyrinG and neurofascin, and loss of paranodal Caspr.32 Electrophysiology showed loss of inward Na+ and outward K+ currents. The pathological abnormalities were prevented by adding complement or calpain inhibitors. The electrophysiological dysfunction was prevented by complement inhibition but not by calpain inhibition. These findings indicate that the pathophysiological process in AMAN is initially characterised by functional axonal involvement manifesting electrophysiologically as CB due to paranodal myelin detachment with node lengthening (figure 3A), Na+ channel disruption (figure 3B) and altered ion and water homeostasis. This process may stop and reverse rapidly, as in AMAN with CB (figure 5B, D), or progress to axonal degeneration (figures 4B and 5C). Conduction slowing in AMAN nerves may be explained by increased node capacitance and partial inactivation of the Na+ channel, which increase the rise time of depolarisation of the action potential and prolong internodal conduction time (figure 5B). The description of patients with AMAN with CB evolving to axonal degeneration or showing reversible CB and axonal degeneration co-occurring in the same or different nerves confirms a pathophysiological continuum.33 This explains why recovery in patients with AMAN may be either very rapid and complete or prolonged with poor outcome, according to the relative amount of CB and axonal degeneration in each patient.
Not considering distal axonal CB may induce, on a single test showing reduced distal CMAP amplitudes interpreted as axonal degeneration, to erroneously formulate a poor prognosis (figure 5D).34 The current electrodiagnostic criteria for GBS do not include CB as an expression of axonal pathology. Therefore, not distinguishing axonal from demyelinating CB by serial recordings might erroneously classify patients with AMAN with CB as acute inflammatory demyelination polyneuropathies and could explain the previously reported association between acute inflammatory demyelinating polyneuropathy and antiganglioside antibodies.21 ,27 In recent studies in Italian and Japanese GBS populations, considering CB as expression of axonal involvement, 24–38% of patients changed classification and the percentages of axonal subtypes were double at follow-up compared to the diagnosis based on the first electrophysiological study.33–35
The above findings explain why AMAN does not fit in the dichotomous classification in demyelinating and axonal neuropathies, and why it is the prototype of nodopathies.
Multifocal motor neuropathy
Multifocal motor neuropathy (MMN) is characterised by slowly progressive, asymmetrical, predominantly distal limb weakness with absent sensory loss, persistent motor CB, presence of IgM to GM1 in about 50% of patients and response to intravenous immunoglobulin in up to 90% of patients.36 In addition to CB, MMN is characterised by axonal degeneration. CB and axonal loss are correlated and independent determinants of each other, suggesting that these processes are linked.37 It has been debated whether MMN is a primary demyelinating or axonal disorder. Pathology studies are scarce and contradictory, with evidence of mild demyelination as well as of primary axonal degeneration.38 ,39 Injection of human sera containing IgM anti-GM1 antibodies into the rat sciatic nerve induces CB, immunoglobulin deposition at the nodes of Ranvier, nodal widening and some paranodal demyelination.40 Application of high concentration anti-GM1 sera to rat single myelinated axons after addition of complement, reduced Na+ currents sufficient to block action potential electrogenesis.41 Very recently, complement activating antibodies to neurofascin and or gliomedin were found in 62% of patients with MMN; interestingly 10% of patients without IgM anti-GM1 reactivity had antineurofascin antibodies.42 The localisation and function of neurofascin and gliomedin (figure 1C) suggest an additional role of antibodies to these axoglial proteins in the involvement of the nodal region in MMN.
Excitability studies performed distal to sites of CB revealed abnormalities similar to those encountered during application of hyperpolarising current, suggesting that axons in MMN are hyperpolarised.43 It was hypothesised that, at the site of the lesion with CB, Na+/K+ pump was blocked because of oedema or the effect of antibodies leading to permanent depolarisation and continuous Na+ influx, possibly through persistent Na+ channels (figure 3C). At adjacent, healthy parts of the axolemma, the accumulated Na+ were suggested to be removed by increased Na+/K+ pump activity, resulting in focal hyperpolarisation (figure 3D). The hyperpolarisation seems to be a stable steady state and this may explain the characteristic persistence of CB. The coexistence at the lesion site of depolarisation and hyperpolarisation could explain the co-occurrence of positive signs such as cramps and fasciculations with predominantly negative features such as weakness and muscle atrophy.
Increased weakness during cold occurs in 83% of patients with MMN.44 This is in contrast with the effect of cooling, which alleviates CB caused by demyelination. It was hypothesised that cold paresis in MMN is not related to demyelination, but to the effect of cold on axons that are focally depolarised but still able to conduct impulses at ambient temperature. The Na+/K+ pump has slower kinetics at low temperatures and cooling would exacerbate the effect of an already compromised Na+/K+ pump, inducing further depolarisation of the membrane and CB.45
In MMN, permanent axonal depolarisation may induce increased Na+ influx, intra-axonal Na+ accumulation, reversal of the axolemmal Na+/Ca2+ exchanger, intra-axonal Ca2+ accumulation and Ca2+-mediated axonal degeneration (figure 4A).
All the above findings suggest that MMN could be better classified as a chronic dysimmune nodopathy.
Other neuropathies with antiganglioside antibodies
Evidence in humans and animal models point to dysfunction/disruption of the nodal region as a common pathogenic mechanism in other acute and chronic neuropathies associated with antibodies to gangliosides. Axonal CB has been described in motor and sensory fibres of patients with acute motor and sensory axonal neuropathy, and the pharyngeal-cervical-brachial subtype of GBS.46 ,47 Axonal CB restricted to sensory fibres has been reported in patients with promptly reversible acute sensory ataxic neuropathy and IgG anti-GD1b or anti-GQ1b antibodies, and in Miller-Fisher syndrome, which is also known to have a good prognosis.48 ,49 The sensory nerve biopsy of an anti-GQ1b ganglioside positive patient with Miller-Fisher syndrome showed lengthening of nodes of Ranvier, myelin splitting and macrophage invasion of the internodal axon without any features of demyelination, strikingly similar to the pathological features found in AMAN motor fibres.50 Chronic ataxic neuropathy patients with disialosyl antibodies may have a relapsing remitting clinical course; relapses have been reported to improve and resolve within a few days with immunotherapy and in some patients persistent multifocal motor CB has been also described.51 All these findings suggest that neuropathies with antiganglioside antibodies belong to a continuous spectrum with a common pathophysiological mechanism focused to the nodal region.52
Critical illness polyneuropathy
Neuropathy occurs in 50–70% of critically ill patients and is thought to be primarily axonal. Some evidence suggests that physiological factors contribute to the pathophysiology. In biopsies performed early after the onset, most sensory nerves were normal, despite the reduced SNAP amplitudes, whereas in late biopsies electrophysiological and histological findings were concordant.53 A nerve excitability study showed findings interpreted as indicative of a chronic membrane depolarisation.54 More recently, critically ill patients who rapidly recovered from the neuropathy with normalisation of previously decreased CMAPs and SNAPs were described.55 This observation prompted the electrophysiological study of a rat model of sepsis. The mixed tail nerve potential was reduced in amplitude after the induction of sepsis but improved in a few days in the surviving animals. Excitability studies showed a hyperpolarised shift in the voltage dependence of Na+ channel inactivation causing increased Na+ channel inactivation.55 Soluble factors released during sepsis might account for the reduction of excitability. For example, nitric oxide, a free radical and inflammatory mediator, induces reversible CB and axonal degeneration in experimental animal models.56 ,57 Electrophysiological and pathological examinations showed that administration of nitric oxide caused disappearance of compound action potential and axonal degeneration that could be prevented by adding Na+ channel or Na+/Ca++ exchanger blockers.58 These findings suggest that inflammation may, through nitric oxide, induce energy restriction by inhibition of mitochondrial activity, consequent Na+/K+ pump malfunction, Na+ influx, axonal depolarisation and failure of conduction (figure 3C). Persistent depolarisation may trigger Ca2+-mediated axonal degeneration (figure 4A).59 The notion that the neuropathy in critically ill patients is due to an altered polarisation of the excitable axolemma at the node of Ranvier and that, to a certain extent, may be reversible, has important implications for prognosis and for the search of timely targeted treatments.
Ischaemic neuropathy
Acute distal multiple mononeuropathies have been rarely described after procedures of arteriovenous shunt in the proximal arm or acute occlusion of a major proximal artery in the extremities. On the other hand, peripheral neuropathy due to ischaemia is often encountered in patients with vasculitis. Nerve biopsies show changes consistent with an axonal disorder. It has been debated whether there is a true CB in ischaemic neuropathies. An abnormal amplitude reduction of proximal CMAP, which usually disappears if nerve conductions are repeated in 2 weeks, occurs in approximately 15% of patients. This finding was thought to be the result of advancing Wallerian degeneration and due to the fact that the distal axons are still able to conduct for a few days after proximal transection. For this reason, the amplitude reduction of proximal CMAP was thought to not represent a true CB and has been called a pseudoCB.60 However, reversible CB has been described in vasculitic and other acute and chronic ischaemic neuropathies.61–64 In these cases, the resolution of CB without development of excessive TD has been thought to be incompatible with segmental demyelination/remyelination. Transient or prolonged CB without TD has been reported following experimental acute occlusion of vasa nervorum by injecting low and high doses of arachidonic acid.65 Morphological examination did not reveal segmental demyelination suggesting a ‘metabolic’ nature for CB. Ischaemia impairs energy-dependent processes and particularly the Na+/K+ pump resulting in accumulation of Na+ ions inside and K+ ions outside the axons with consequent depolarising CB, which may be persistent in chronic ischaemia and resolve after recanalisation (figure 3C).64 ,66 If Na+ influx is excessive, the Na+/Ca2+ exchanger reverts its activity and Ca2+ accumulation in the axoplasma triggers axonal degeneration (figure 4). Therefore, acute ischaemic neuropathies have the characteristic of a nodopathy due to energy failure. It is probably more correct to define the very early reduction of proximal CMAP amplitude as a true axonal CB that rarely is reversible and more frequently progresses to axonal degeneration.
Beriberi neuropathy
An example of nutritional nodopathy, possibly due to energy failure, could be the neuropathy in thiamine deficiency, which has been thought to be axonal in nature.67 In patients with Wernicke-Korsakoff, encephalopathy and polyneuropathy CMAP amplitudes were markedly reduced, and SAPs were not recordable or reduced in amplitude with minimal conduction slowing.68 Sural nerve biopsy showed mild axonal degeneration. Thiamine infusion induced, in 2—4 weeks, normalisation of CMAPs and SAPs, in parallel with clinical improvement of neuropathic symptoms and signs. Thiamine deficiency decreases pyruvate dehydrogenase activity and ATP. This may reduce the Na+/K+ pump activity, inducing membrane depolarisation and conduction failure, and eventually trigger Ca2+-mediated axonal degeneration (figures 3C and 4).
Tetrodotoxin poisoning
Tetrodotoxin (TTX) is contained in the skin, liver and gonads of the puffer fish, which is eaten as a delicacy in Japan. When the fish is not professionally processed and contains too much TTX, it causes, besides the wanted perioral paraesthesias, distal limb paraesthesias, numbness, bulbar weakness and, in severe poisoning, flaccid tetraplaegia with rapid progression to respiratory insufficiency and cardiovascular involvement. In Japan, from 1965 to 2007, 1032 cases of poisoning with 20% of fatalities were reported.69 There is no antidote, but by careful monitoring and appropriate assistance, most moderate and severe cases resolve in 1 week. TTX occludes the outer pore of the Na+ channel affecting the action current at the Ranvier node. Nerve conduction studies show reduced amplitude CMAPs and SNAPs with slow conduction velocities, prolongation of distal motor latencies and F wave. CMAPs, SNAPs and conduction velocities revert to normal in a few days without the development of excessive TD.70 Focal CB in the intermediate nerve segments has not been described in intoxicated patients but TTX, when intraneurally injected in the rat, induces an immediate CB that resolves in a few hours without development of TD.71 Excitability studies showed a higher threshold in motor and sensory axons in patients, and indicate a reduction of Na+ conductance confirming that TTX blocks Na+ channels.72 Partial inactivation of Na+ channels at nodes may increase the rise time of depolarisation of action potential, prolonging internodal conduction time and explains conduction slowing as hypothesised for local anaesthetics.24 The lack of excessive TD is probably due to the fact that both conduction slowing and conduction recovery are quite uniform among nodes and fibres. The transiently reduced distal CMAP amplitudes are due to CB in terminal axons that, because of distal branching, have a reduced safety factor of conduction. TTX poisoning seems to be a good example of a purely functional toxic nodopathy (figure 3B).
Conclusions
The nodopathies of the peripheral nerve are characterised by the following key features:
They have different aetiologies but a final common pathophysiological mechanism characterised by dysfunction/disruption of the excitable axolemma at the nodal region.
They display a pathophysiological continuum from transitory CB to axonal degeneration.
CB may be due to paranodal myelin detachment and node lengthening, dysfunction or disruption of Na+ channels, altered homeostasis of water and ions, and abnormal segmental polarisation of the axolemma.
CB may be promptly reversible without development of excessive TD characteristic of de-remyelination, or may be persistent.
Axonal degeneration, depending on the specific disorder and its severity, eventually follows CB.
In clinical practice, electrodiagnosis can be made by serial conduction studies that document, in acute disorders, a promptly reversible CB or conduction slowing without development of excessive TD or a progression from CB to axonal degeneration. In chronic disorders persistent CB and signs of axonal degeneration may coexist.
Some nodopathies, independently from the aetiology, are ascribable to energy depletion. The nodal region is characterised by increased mitochondria density and high metabolic demand to sustain nerve activity.18 An energetic crisis, through reduced activity of the ATP-dependent Na+/K+ pump, induces depolarisation of nodal membrane and CB, which may be potentially rapidly reversible. If the lack of energy persists, axonal degeneration, possibly due to Ca2+ overload and Ca2+-dependent calpain activation, develops.
The neuropathies we review can be classified with difficulty as axonal or demyelinating. Most of them have been classified as axonal on the basis of pathology. However, all of them present CB that can be reversible or progress to axonal degeneration. In common neurological knowledge the adjective axonal is associated with degeneration and poor prognosis and not with reversible CB and prompt recovery. On the other hand, the finding of CB may classify these neuropathies as demyelinating on the basis of a single electrophysiological study. We deem that the category of nodopathies better reflects the current understanding of the pathophysiology and better systematises the disorders we reported than the traditional classification. The advantages are that the nodopathy term:
points out directly to the site of nerve injury;
circumvents the apparent paradox that something axonal may be reversible and have a good prognosis;
avoids the confusing situation, as in neuropathies associated with antiganglioside antibodies, that in spite of the common site of nerve damage and pathophysiological mechanism, some patients might be classified as having a demyelinating, and others as having an axonal, neuropathy;
emphasises the potential reversibility in neuropathies traditionally thought to be characterised only by axonal degeneration, opening a therapeutic window and stimulating research for timely targeted treatments.
References
Footnotes
Contributors AU contributed with manuscript conception, literature search and wrote the first draft. SK contributed to literature search and critical review of each draft.
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.