Article Text

Abstract
Early aetiological diagnosis is of paramount importance for childhood dystonia because some of the possible underlying conditions are treatable. Numerous genetic and non-genetic causes have been reported, and diagnostic workup is often challenging, time consuming and costly. Recently, a paradigm shift has occurred in molecular genetic diagnostics, with next-generation sequencing techniques now allowing us to analyse hundreds of genes simultaneously. To ensure that patients benefit from these new techniques, adaptation of current diagnostic strategies is needed. On the basis of a systematic literature review of dystonia with onset in childhood or adolescence, we propose a novel diagnostic strategy with the aim of helping clinicians determine which patients may benefit by applying these new genetic techniques and which patients first require other investigations. We also provide an up-to-date list of candidate genes for a dystonia gene panel, based on a detailed literature search up to 20 October 2014. While new genetic techniques are certainly not a panacea, possible advantages of our proposed strategy include earlier diagnosis and avoidance of unnecessary investigations. It will therefore shorten the time of uncertainty for patients and their families awaiting a definite diagnosis.
- DYSTONIA
- GENETICS
Statistics from Altmetric.com
Introduction
Dystonia is a movement disorder characterised by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures or both.1 For dystonia in children and adolescents, here referred to as dystonia of childhood (DC), the list of possible genetic and non-genetic causes is extensive.2 ,3 For clinicians encountering a young patient with dystonia, an important practical question is how to manage the diagnostic workup, which is often challenging, time consuming and costly.
Recently, a paradigm shift has occurred in molecular genetic diagnostics, with next-generation sequencing (NGS) techniques now allowing us to analyse hundreds of genes simultaneously. NGS diagnostic strategies are particularly effective in heterogeneous conditions, including movement disorders, significantly increasing the diagnostic yield at lower costs.4 ,5 As a significant proportion of DC cases is estimated to be genetic, a ‘genetics first’ diagnostic approach for all patients with DC seems logical and appealing. However, there are two groups of patients for whom another initial approach should be considered. First, in children and adolescents who may have acquired dystonia, and second, in patients in whom the cause may be a treatable inborn error of metabolism (IEM), because for most of these IEMs biochemical investigations will be a faster diagnostic method than genetic testing.
We first provide a systematic literature review of the phenomenology, classification and aetiology of DC. We then propose a novel diagnostic strategy that will help clinicians determine which patients may benefit from NGS technologies and which patients require other initial investigations. Finally, we give an up-to-date list of dystonia gene candidates to enhance the development of NGS diagnostics for DC (see online supplement 1).
Methods
We systematically reviewed all papers regarding DC up to 20 October 2014, both genetic and non-genetic, in three age groups (infancy, childhood and adolescence), as proposed in the latest dystonia classification.1 For details of our systematic search, see online supplement 2.
Dystonia in children and adolescents: state of the art
Phenomenology: Is it dystonia?
The first step in diagnosing DC is the identification of a hyperkinetic movement as being ‘dystonic’. Dystonia is defined as “a movement disorder characterised by sustained or intermittent muscle contractions causing abnormal, often repetitive, movements, postures or both. Dystonic movements are typically patterned or twisting, and may be tremulous. They are often initiated or worsened by voluntary action and associated with overflow muscle activation”.1 This definition of dystonia is identical for adults and children1 ,3 and similar to the definition of dystonia published by the Taskforce on Childhood Movement Disorders.6 In children, dystonia is more often generalised compared with adult-onset dystonia.
Correct identification of dystonia involves both an understanding of classification systems and visual pattern recognition. Three important, characteristic, clinical features of dystonia are: (1) patterned, predictable contractions of the same muscles; (2) exacerbation when performing voluntary movements (eg, walking, running, writing) and (3) the so-called geste antagoniste, or sensory trick. This phenomenon is characterised by the relief of dystonic movements by lightly touching the relevant or adjacent part of the body. A sensory trick is particularly frequent in cranial and cervical dystonia, whereas limb and trunk involvement more often predominate in children. Therefore, a sensory trick is not an obligatory feature in DC; however, when observed, it strongly favours a diagnosis of dystonia.1 ,6
In children, movements should be evaluated in relation to their developmental age. For instance, a healthy toddler can have normal overflow movements that may look like dystonia, diminishing as the child's development progresses.3 In addition to these normal movements, abnormal movements may also mimic dystonia (table 1). For example, children with focal, stereotyped movements of the eyelids, face or neck are more likely to have tics than focal dystonia.7 ,8
Mimics of dystonia in children and adolescents
Reliable diagnostic criteria for different body localisations of dystonia are needed to help clinicians accurately differentiate dystonia from conditions mimicking dystonia. Recently, a diagnostic guideline for diagnosing blepharospasm has been validated;9 however, blepharospasm is a form of focal dystonia that rarely occurs in childhood or adolescence. For other body localisations of dystonia, specific diagnostic criteria are an unmet need.
Classification of dystonia
The most recent general classification scheme of dystonia identifies two distinct axes: axis I—clinical characteristics, and axis II—aetiology.1 Axis I describes the clinical features by (1) age at onset, (2) body distribution, (3) temporal pattern, (4) coexistence of other movement disorders and (5) other neurological or systemic manifestations. Axis II addresses the aetiology via two components: (1) nervous system pathology and (2) whether the dystonia is inherited or acquired. Classification of aetiology into the categories ‘inherited’ or ‘acquired’ differs from traditional classification schemes in which dystonia was classified into primary genetic dystonia or secondary dystonia.1 The reason for this change was that primary dystonias, heredodegenerative dystonias and dystonia-plus syndromes are all in fact genetic disorders.1 These three categories are now considered together as ‘inherited’. In this review, we elaborate on this recent change in aetiological classification.
Aetiology of dystonia
There are many possible aetiologies of DC. For this review, we highlight acquired dystonias and treatable IEMs because an initial approach other than NGS testing needs to be considered for these conditions. All other genetic causes can be tested at the same time by means of NGS diagnostics.
Acquired dystonias
We focus on acquired forms of dystonia that are relatively common and/or treatable. Drugs and toxic agents that may cause DC are listed in table 2. For other causes of acquired DC, clinical clues and recommended investigations are summarised in table 3.
Drugs and toxic agents that may cause dystonia in children and adolescents
Clinical clues suggesting acquired dystonia
Drugs and toxic agents
DC can be induced by certain drugs and toxic agents, most commonly neuroleptics and antiemetics (table 2).7 ,8 Drug-induced dystonias are categorised into acute dystonic reactions and tardive (chronic use) dystonia. The latter is a well-recognised disorder in adults, but may also occur in children.7 Acute forms of dystonia may arise after taking a few doses or even after one administration or accidental ingestion.8 The dystonia usually disappears rapidly on withdrawing the offending drug.
Cerebral palsy
Dyskinetic cerebral palsy (CP) is the most common cause of acquired DC.10 CP is a clinical diagnosis, encompassing a group of permanent disorders that cause impairment of movement and posture, attributed to non-progressive disturbances that occurred in the developing fetal or infant brain.11 Dyskinetic CP is characterised by the presence of choreoathetosis and dystonia11 and possible aetiologies are heterogeneous.8 ,12 It is most common in children, born at term, who have experienced adverse perinatal effects, since the basal ganglia are particularly vulnerable to pathogenic events towards the end of gestation.12 There are guidelines to help identify whether an acute intrapartum event was the likely cause of any particular case of CP.13 Owing to the aggressive treatment of perinatal hyperbilirubinaemia, it is now rare to see kernicterus as a cause of dyskinetic CP.12
In dyskinetic CP, the hyperkinetic movements are usually bilateral and mostly begin after the first year of life, and progress slowly for several years.7 ,8 In children with severe CP, dystonia may be so profound and sustained that it manifests as hypertonia rather than abnormal involuntary movements.3 Brain MRI demonstrates abnormal findings in about 80% of individuals with CP.14 Genetic analysis is recommended in those cases where no specific cause can be determined, as several monogenic disorders can present with clinical features similar to CP.15
Acquired structural lesions
Structural lesions, such as stroke, neoplasms or structurally abnormal vessels including arteriovenous malformations, may result in unilateral DC (focal or hemidystonia).7 ,8 Childhood stroke may result in dystonia if the caudate, lenticular nucleus or thalamus are involved.7 ,8 In most cases, the dystonia develops months or even years after the incident.
Autoantibody-associated and autoimmune disorders
Several autoantibody-associated and autoimmune disorders can lead to DC (table 3).16 We put emphasis on two autoantibody-associated disorders, as early recognition and timely therapy can improve the outcome significantly in these conditions.16
Anti-N-methyl-d-aspartate receptor (NMDAR) encephalitis in children is characterised by a combination of seizures, movement disorders, psychiatric symptoms and encephalopathy.16 The first symptom is often non-psychiatric.17 In addition to dystonia, multiple movement disorders can be seen in the same patient,16 the most characteristic being orofacial dyskinesias.17 Young children often present with temper tantrums, hyperactivity or irritability, whereas in older patients anxiety, psychosis and altered personality are the main psychiatric features observed.17 Recognition of the combination of symptoms should prompt testing for anti-NMDAR antibodies, both in serum and cerebrospinal fluid (CSF).17 Brain MRI, EEG and CSF may all show non-specific abnormalities.17 ,18 An underlying neoplasm is found in approximately 6% of girls younger than 12 years but rarely in boys, whereas the association with an ovarian teratoma increases in adolescent girls.18 Treatment consists of immunotherapy and oncological treatment in those patients with a clinically detectable tumour.18 Outcome is good in the majority of patients treated early enough.18
Autoimmune basal ganglia encephalitis is a syndrome characterised by extrapyramidal movement disorders including dystonia and parkinsonism, sleep disturbance, dysautonomia and psychiatric symptoms.16 Approximately 70% of cases have serum antidopamine-2 receptor antibodies.16 Many patients have MRI T2 hyperintense basal ganglia abnormalities and show signs of CSF inflammation including oligoclonal bands.16 Immune therapy is the mainstay of treatment.16 ,17 In the past, encephalitis with dominant involvement of the basal ganglia was given a variety of names, including encephalitis lethargica and (infantile) bilateral striatal necrosis.16 These disorders and autoimmune basal ganglia encephalitis may all be part of the same clinical entity.16
Infections
DC caused by infection is relatively rare, but has been reported in children with viral infections, tuberculosis, mycoplasma or toxoplasmosis.19 Infection by flaviviruses is an important cause of DC, the most common being Japanese encephalitis.19 Other viruses associated with DC include influenza viruses, herpes viruses (including herpes simplex and herpes zoster) and measles viruses, which may lead to subacute sclerosing panencephalitis.7 ,8 The main bacterial infections are tuberculosis and infection by Mycoplasma pneumoniae.8 Infection should be suspected in any child with dystonia and pre-existing immunodeficiency or signs of meningoencephalitis or encephalitis. Detecting the infectious agent may be important for the type of therapy chosen, and therefore serum and CSF investigations are indicated in addition to neuroimaging.
Treatable IEMs
IEMs are highly heterogeneous. For most clinicians who do not work daily with IEMs, it will be virtually impossible to recognise all these often extremely rare conditions. Fortunately, since all IEMs can be detected with NGS diagnostics, early identification is only necessary for those IEMs where timely treatment can improve the outcome.20
In general, an important clue for an IEM is a complex clinical picture comprising both neurological and non-neurological features. An overview of treatable IEMs associated with DC is provided in online supplement 3. We defined ‘treatable’ as the availability of a therapy that might lead to the improvement or prevention of symptoms. We will highlight five significant subgroups of treatable IEMs that may cause DC.
Organic acidurias
Organic acidurias can present both acutely and intermittently and are associated with ‘intoxication-like’ non-specific symptoms, such as vomiting and anorexia, progressing towards encephalopathy. Episodes are frequently triggered by intercurrent illness, dietary changes or prolonged fasting.21 When the underlying enzymatic defect is severe, onset will be in the newborn period. Milder phenotypes may present later as a slowly progressive disorder or with an intermittent course. Examples of organic acidurias associated with DC are propionic aciduria, methylmalonic aciduria, cobalamin defects and glutaric aciduria type I.22
GLUT-1 deficiency
GLUT-1 deficiency, caused by mutations in the SCL2A1 gene, can give rise to paroxysmal dystonia triggered by prolonged exercise.23 This phenotype is also referred to as paroxysmal exertion-induced dystonia. The SCL2A1 gene encodes for the glucose transport protein 1, and mutations in this gene compromise glucose transport to the brain. Paroxysmal dystonia can be the sole feature, but developmental delay, spasticity, ataxia and epilepsy can also be part of the phenotype. A ketogenic diet is the current gold standard for treatment and has proven to be beneficial in most cases.23
Metal storage
Wilson's disease (WD) and dystonia with brain manganese accumulation (DBMA), caused by SLC30A10 mutations, are both metal storage disorders in which symptoms can be fully or partly prevented by timely treatment.24 ,25 In both disorders, a combination of neurological symptoms and hepatic involvement is usually present. Other manifestations are psychiatric symptoms and a corneal Kayser-Fleischer ring in WD and parkinsonism and polycythaemia in DBMA. Indicative biochemical findings include low serum copper and ceruloplasmin in WD and hypermanganesaemia in DBMA.
Lysosomal storage
Niemann Pick type C is a clinically heterogeneous disorder in which the presenting phenotype depends on the age of onset. Infants can present with ascites and liver or pulmonary disease. The classic presentation in mid to late childhood consists of ataxia, a supranuclear vertical gaze palsy, psychiatric symptoms, dystonia and dementia, whereas the clinical picture in adults is dominated by psychiatric symptoms and cognitive decline.26 Recently, treatment with miglustat has been shown to stabilise the progression of neurological symptoms, including in paediatric patients.27
Dopa-responsive dystonias
Dopa-responsive dystonias (DRD) are a group of disorders with a more insidious onset, probably representing 5% of childhood dystonias.28 The autosomal dominant form, GTP-cyclohydrolase deficiency, is most common. This form is also known as Segawa's disease and shows an excellent and sustained response to low doses of levodopa.29 Typically, there is a diurnal fluctuation of symptoms, and associated parkinsonism. Furthermore, two autosomal recessive forms of DRD have been identified: tyrosine hydroxylase deficiency and sepiapterin reductase deficiency, both often accompanied by intellectual disability and ophthalmological problems like oculogyric crisis, upward gaze and ptosis.30
Since DRD features can be non-specific and can show considerable phenotypic variability, DRDs are frequently misdiagnosed as CP.30 This may result in a considerable delay in diagnosis and adequate treatment.29 ,30
In addition to biochemical and molecular studies, a levodopa trial can be used as a diagnostic procedure. However, it should be noted that a positive response on a levodopa trial is not specific for the classic DRDs, but can also be seen in other disorders such as ataxia telangiectasia and GLUT-1 deficiency.31 ,32
Classification of genetic dystonias
The genetic forms of dystonia including IEMs may be categorised into two groups. The first group consists of the monogenetic forms of dystonia with assigned genetic loci identified as DYT1–25, formerly named ‘primary dystonias’ and ‘dystonia plus syndromes’. These disorders are characterised by isolated dystonia, or dystonia combined with parkinsonism or myoclonus.1 The second group consists of genetic disorders in which dystonia is an important feature among several other neurological and systemic features. On axis I of the latest dystonia classification, these co-occurring neurological or systemic manifestations are classified as ‘associated features’.1 Important associated features in children include: ataxia, epilepsy, mental retardation, spasticity, hypotonia, abnormal eye movements, neuropathy, deafness, ophthalmological signs, hepatosplenomegaly, psychiatric and dysmorphic features. These features are decisive for accurate phenotyping and a prerequisite for correct interpretation of NGS results.
NGS methodology
Genetic techniques using massive parallel sequencing are called NGS. With these new techniques, sequencing the entire genome of a patient (whole-genome sequencing; WGS), the coding regions (exons) of every gene (whole-exome sequencing; WES) or targeting specific disease-causing genes (targeted resequencing; TRS) have all become a reality in DNA diagnostics. Technical details of the specific methods fall outside the scope of this review, but are described elsewhere.33
It is important to recognise that with WGS or WES approaches, information for all genes will become available, including those not relevant to the diagnostic question. These genes need to be excluded to restrict the data analysis to a list of known genes that might explain the phenotype. If the phenotype is unique and no mutation is found in the selected genes, the information about the excluded genes may be used to hunt for new disease-causing genes. The drawbacks of WGS and WES are high costs, the risk of unsolicited findings, and coverage that is usually less than in TRS panels, compromising the diagnostic accuracy. In TRS panels, a preselected list of several known genes that cause dystonia are tested. By sequencing only preselected genes, the coverage increases significantly, contributing to diagnostic accuracy, and unsolicited findings are minimised, at significantly lower costs.
The important benefits of NGS diagnostics compared with regular biochemical procedures are that shipping DNA to referral centres is relatively cheap and straightforward, without stringent shipping conditions. In contrast, the costs and conditions of shipping samples, for instance, for (CSF) biochemical tests can be a serious hurdle in the present diagnostic process.
It is to be expected that in the near future the widespread use of NGS, both in research and in clinical diagnostics, will lead to many more reports of dystonia-associated genes, and the list of associated genes will grow rapidly. However, it is important that independent confirmation of the causal relationship between gene variants and dystonia is performed because, in some of the recently annotated dystonia genes, variants in these genes also occur with high frequency in the general population.34
A new diagnostic algorithm
Owing to the extraordinarily broad range of possible causes of DC, several algorithms have been developed to assist clinicians in making diagnostic decisions.2 ,35 ,36 These algorithms are not widely applicable as they mainly focus on (rare) neurometabolic causes and do not make use of the availability of NGS methodologies. On the basis of our systematic literature review and our own clinical experience, we propose a new diagnostic algorithm with five steps (figure 1).
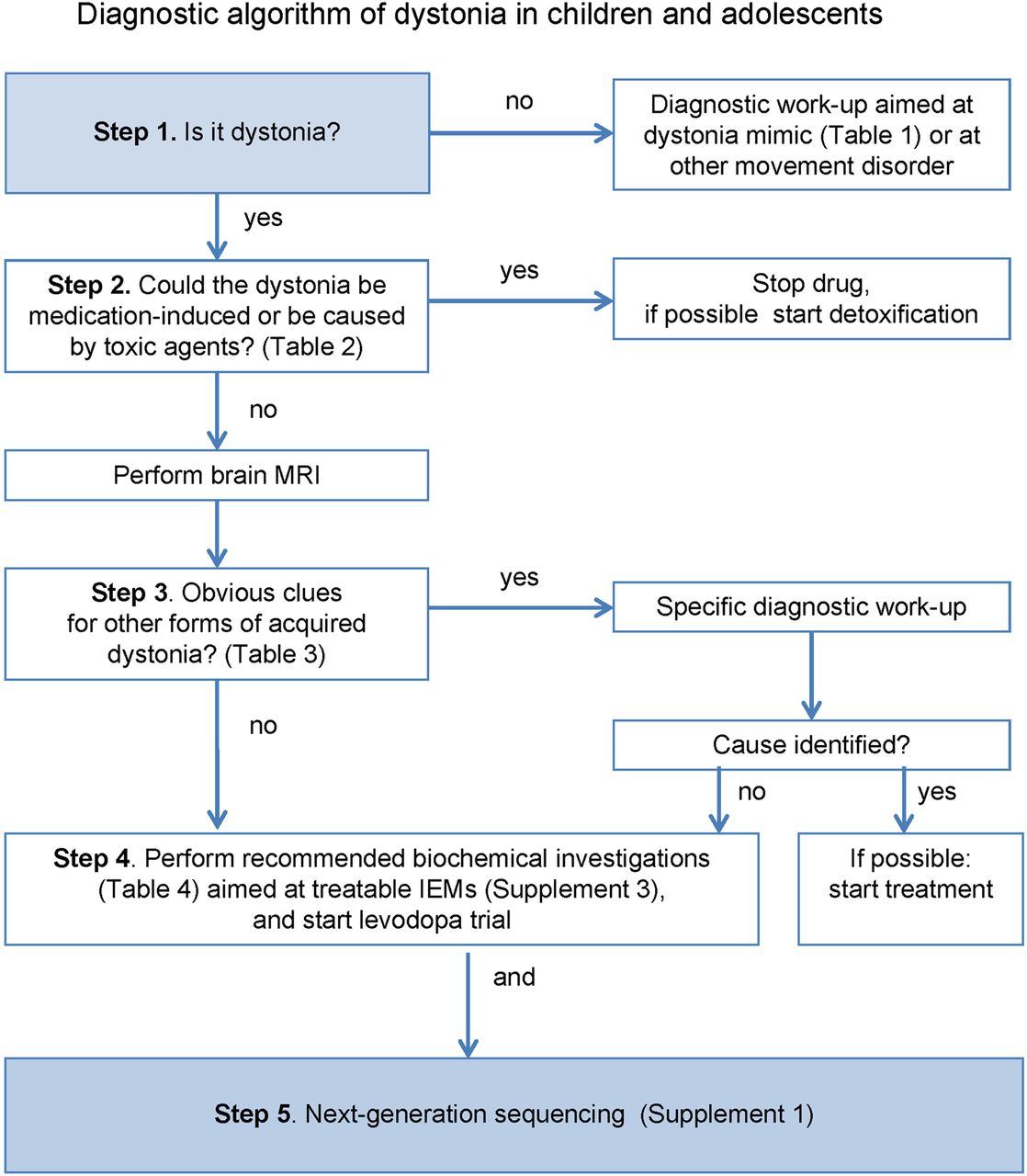
{kind=link}
Diagnostic algorithm of dystonia in children and adolescents (IEM, inborn error of metabolism).
Step 1: Is it dystonia?
The first step in the algorithm is to record a careful history and perform a physical and neurological examination to determine that dystonia is an important feature.
Movement disorders that may be misdiagnosed as dystonia are listed in table 1. In general, these ‘pseudodystonias’ have a known or presumed cause that is thought to differ from the causes of the broader dystonia group.1 Applying the algorithm and using NGS testing is not advised in these conditions.
Step 2: Could the dystonia be medication-induced or caused by toxic agents?
The second step is to verify exposure to any medication or toxic agents that could be causing the dystonia (table 2). Treatment consists of discontinuing medication or prevention of further toxic exposure and, if possible, detoxification.
Step 3: Clinical clues suggesting acquired dystonia?
Step 3 is to consider whether the dystonia could be acquired. In table 3, we indicate red flags for acquired disorders with the main subgroups. These red flags are only defined to guide clinicians to a limited number of disorders in which immediate diagnosis and treatment is necessary to identify treatable disorders, preventing insults to the brain during the diagnostic process.
Step 4: Biochemical investigations and levodopa trial
In any child with dystonia without obvious clues for an acquired cause, we recommend performing a laboratory workup (table 4) aimed at identifying the treatable forms, before moving on to NGS testing. Of course, this recommendation only applies for those centres where biochemical diagnostics will provide faster results than NGS testing, depending on the local facilities. CSF investigations are only recommended in selected patients (table 4) because otherwise the diagnostic yield of CSF investigations is likely to be rather low.37 ,38
Biochemical investigations to identify treatable inborn errors of metabolism with dystonia as an important feature
In addition to the laboratory investigations, we recommend that all patients receive a trial of levodopa with carbidopa.30 The primary goal of the trial is diagnostic. However, an additional advantage is that levodopa can also give symptom relief in non-DRD dystonia.39 The recommended starting dose of levodopa is 1 mg/kg/day, to be gradually increased until complete benefit, or until dose-limiting side effects occur.7 Most individuals respond to 4–5 mg/kg/day in divided doses.40 Levodopa should be given for 3 months before considering the trial a failure.39
Step 5: NGS
Simultaneously with the biochemical investigations and the initiation of the levodopa trial, all possible genetic causes can be approached by using NGS diagnostic technologies. To facilitate this, we provide a list of DC-associated genes (see online supplement 1). For those cases that remain unsolved after NGS testing, referral to a tertiary referral centre is recommended to further explore the possibilities to obtain an aetiological diagnosis.
Discussion
We provide a comprehensive overview of DC and propose a new diagnostic algorithm (figure 1). This five-step approach provides guidance for clinicians to determine which patients may benefit from innovative genetic tests and those for whom other investigations are required first, while taking into account the importance of early recognition of acquired and treatable causes of DC.
Our proposed flow chart (figure 1) differs from existing algorithms in that certain commonly used processing steps have been omitted, such as age at onset, temporal pattern (eg, persistent or paroxysmal), associated features and mode of inheritance.2 ,35 ,36 Indeed, ‘pattern recognition’ based on these features has been important in the delineation of dystonia disorders and can still be successful in identifying classical phenotypes, especially by experts in the field.1 ,8 However, these features were not included in our algorithm because many clinicians will have limited experience with these rare disorders and specific clinical patterns will easily remain unrecognised. In addition, recent insights from more widely applied NGS testing demonstrate that the clinical heterogeneity of many disorders is much larger than expected,23 ,31 so clinical pattern recognition of milder, intermediate and unusual phenotypes remains problematic.
Nevertheless, careful clinical phenotyping still remains indispensable for two reasons. First, clinicians need to define, on the basis of these clinical parameters, the a priori risk that the patient is indeed suffering from a genetic disorder. NGS methodology should not be used when the a priori risk is low, because the numerous genes being tested increase the chance that variants will be misinterpreted as disease-causing, in genes that are unlikely to explain the clinical phenotype. Second, closely related to the first reason, detailed phenotyping is key when the results of NGS diagnostic strategies are available and need to be interpreted. As Hennekam and Biesecker41 clearly stated, NGS and computers will not magically make patient diagnoses for us. Instead, there will be a shift from a pre-NGS-test differential diagnostic mode to a post-NGS-test diagnostic assessment mode.41 Thus, the diagnostic skills of clinicians will be integrated into the evaluation of NGS test results to make molecular diagnoses together with laboratory staff.
Notably, clinicians using NGS diagnostics should be aware that there are some technical pitfalls in the application of NGS diagnostics such as a limited ability to detect large structural rearrangements. In DC, this is particularly relevant if no causative mutation in a gene can be identified by NGS techniques, while at the same time the clinical picture is compatible with, for example, myoclonus dystonia or paroxysmal kinesigenic dyskinesia, both disorders that may be caused by deletions (in SCGE and PRRT2, respectively). In these cases, additional genetic tests detecting deletions are still required, such as multiplex ligation-dependent probe amplification or array-comparative genomic hybridisation (array-CGH).42
At present, we live in a period of transition between emerging NGS diagnostic tests and changing costs, budgets and availability of diagnostic procedures. In the future, NGS tools will become increasingly available in many areas of clinical diagnostics and clinical decision-making, and will be incorporated in our daily work and change our daily routines. Although not a panacea, the advantages of this new strategy will be earlier diagnosis, avoidance of unnecessary investigations and the possibility of genetic counselling for family members. It will crucially shorten the time patients with DC and their families spend in uncertainty awaiting a definitive diagnosis.
Acknowledgments
The authors thank Kate McIntyre and Jackie Senior, University Medical Center Groningen, Department of Genetics, for editing the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online supplement
- Data supplement 3 - Online supplement
- Data supplement 4 - Online supplement
Footnotes
Contributors MEvE was involved in the design and conceptualisation of the study, analysis and interpretation of the data, drafting and revision of the manuscript. AK was involved in the analysis and interpretation of the data, drafting and revision of the manuscript. HE, RJS, CCV-B, DAS and OFB were involved in the revision of the manuscript. MAJT and TJdK were involved in the design and conceptualisation of the study, analysis and interpretation of the data, and revision of the manuscript.
Competing interests MAJT received research grants from Fonds Nuts-Ohra, Stichting wetenschapsfonds dystonie vereniging, Prinses Beatrix Foundation, STW Technology society (NeuroSIPE). Unrestricted grants were received from Ipsen Pharmaceuticals, Allergan Pharmaceuticals and Medtronic for a dystonia nurse, DystonieNet and a teaching course. TJdK received a research grant from Metakids foundation.
Provenance and peer review Not commissioned; externally peer reviewed.