Article Text

Abstract
While the past 2 decades have witnessed an increasing understanding of amyotrophic lateral sclerosis (ALS) arising from East Asia, particularly Japan, South Korea, Taiwan and China, knowledge of ALS throughout the whole of Asia remains limited. Asia represents >50% of the world population, making it host to the largest patient cohort of ALS. Furthermore, Asia represents a diverse population in terms of ethnic, social and cultural backgrounds. In this review, an overview is presented that covers what is currently known of ALS in Asia from basic epidemiology and genetic influences, through to disease characteristics including atypical phenotypes which manifest a predilection for Asians. With the recent establishment of the Pan-Asian Consortium for Treatment and Research in ALS to facilitate collaborations between clinicians and researchers across the region, it is anticipated that Asia and the Pacific will contribute to unravelling the uncertainties in ALS.
- ALS
- MOTOR NEURON DISEASE
- EPIDEMIOLOGY
Statistics from Altmetric.com
Introduction
Amyotrophic lateral sclerosis (ALS) is characterised by degeneration of the upper and lower motor neuronal systems, heralded by progressive muscle weakness, paralysis and death.1 Although there exists a growing body of ALS research that has largely originated from developed Western countries, the Asian region represents the majority of the world population. While recent years have seen a rise in ALS research from Asia, knowledge of the disease in Asia, especially from less developed countries, remains limited. Asians represent a widely diverse population in ethnic, social and cultural background as well as in the provision of healthcare.
Studies from mixed populations suggest phenotypic variability between patients from different ethnic backgrounds, although true differences remain a source of ongoing debate.2 Recent discoveries of new genes in ALS have led to greater insight into the underlying pathophysiology of motor degeneration. The C9orf72 repeat expansion accounts for a significant percentage of familial and sporadic ALS in Caucasian populations but is rare in Asian cohorts.3 Speculative observation suggests that the origin of this mutation dates back 1500 years to the Vikings, relating its spread to other populations through their European invasion.4 ,5 It remains to be determined whether a similar founder effect exists in Asia and within other ethnic groups.
In this review, current knowledge of ALS in Asia will be presented, highlighting similarities and differences with other disease populations. Central to addressing the current shortcomings, there is a clear need to establish an active collaborative network of clinicians and researchers within the region and its surrounding partners that can work in synergy to achieve greater insight into this devastating disease.
Epidemiology
Western population-based studies have suggested a uniform incidence rate of ALS at 2.16 per 100 000 person-years.1 There is evidence to suggest that in populations where there is genetic admixture, the incidence of ALS is lower.6 This suggests that heterogeneous populations are less likely to share ‘at-risk’ genes that may result in increased susceptibility of developing ALS. Asia represents a large geographical area with populations from diverse ancestral backgrounds and within these populations, there are likely to be inter-racial unions resulting in a large genetic and racial admixed population. For this reason, population studies in Asian cohorts are of interest. Although the reported prevalence and incidence of ALS from different Asian countries exists, most have been extrapolated from single centred or multicentred cohort studies, rather than true population-based studies, thereby introducing significant bias (table 1 and figure 1). In Japan, the crude incidence7 is similar to that in western cohorts, while a lower incidence is observed in Chinese patients.8 ,9 A house-to-house population survey in India showed a prevalence of 4/100 000, inferring a low incidence.10 The reasons underlying the low incidence in many Asian cohorts are less certain. Possibilities include the low incidence of genetic mutations in ALS such as the C9orf72 repeat expansion and the lower life expectancies in less-developed Asian countries.
Demographic data of patients with ALS from various Asia-Pacific countries based on population-based and cohort studies
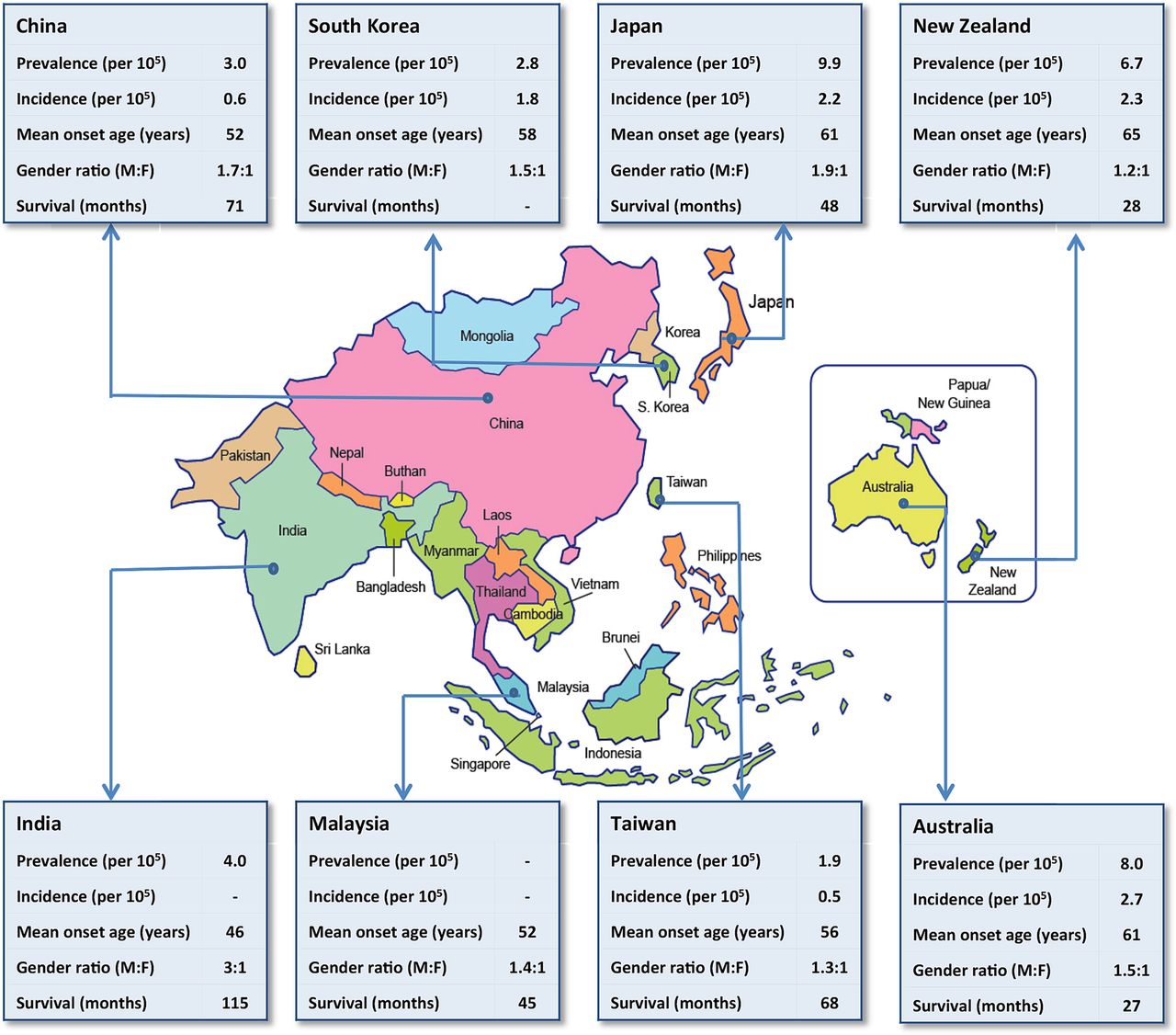
Representative demographic data concerning amyotrophic lateral sclerosis of some of the countries in the Asia-Pacific region (refer to table 1 for source data). F, female; M, male.
In Japan and South Korea, the mean age of disease onset is 61 and 58 years, respectively,11 ,12 but other Asian cohorts have suggested a younger age of disease onset compared with Caucasians. In China, the mean age is approximately 52 years,13–15 whereas in a single study from India, the age of onset was 46 years.16 The overall gender ratio of Asian patients with ALS is comparable to that of Caucasian patients where there is a higher occurrence in males compared with females of between 1.5 and 1.7:1.17 ,18 In Indians, however, there appears to be an even higher proportion of males affected, but it would be difficult to ascertain if this is a true reflection of the disease pattern or referral bias as existing reports have come from clinic-based studies rather than population-based studies.16 ,19 Familial patterns have been reported to account for 5–10% of patients with ALS.1 In Asia, studies to date suggest that familial ALS is <5%.12 ,13 ,16 ,19 ,20
Genetic perspectives
At present, >20 genes have been implicated or associated with disease susceptibility in ALS.21 Table 2 depicts the frequencies of mutations in common ALS-causing genes from the different Asian countries in familial and sporadic ALS. We now know that the most common genetic mutation in the Caucasian population is C9orf72, which accounts for >40% of familial and 5–20% of sporadic ALS.22 Reports from Asian cohorts have suggested that this mutation occurs less frequently.20 ,23–27 In 563 Japanese patients with ALS, C9orf72 accounted for only 2 patients (0.4% sporadic ALS; 0% familial ALS). In one patient, there was an overlap with frontotemporal dementia (FTD) and other family members had either dementia or primary progressive aphasia. In another patient, an asymptomatic sibling aged 76 years with the mutation supported the theory of non-penetrance that has been described elsewhere.5 The common sharing of a shorter region of the Finnish risk haplotype is likely to reflect the migration patterns from the West to East Asia.5 ,20
The frequencies of mutations in common ALS-causing genes from different Asian cohorts
In a recent study utilising next generation sequencing in 508 Japanese patients, mutations in SOD1 accounted for the most frequent mutations in familial (36%) and sporadic ALS (2.3%).28 This is similarly seen in mainland China where the most frequent mutations in familial ALS are in SOD1 (27.9%), FUS (7.1%), TARDBP (3%) and C9orf72 (2.2%). In sporadic ALS, mutations in FUS (1.9%) are most common, followed by SOD1 (1.3%), TARDBP (0.5%), C9orf72 (0.4%) and ANG (0.3%).29 Interestingly, a separate haplotype analysis of single-nucleotide polymorphisms (SNPs) within or surrounding C9orf72 in two Chinese patients with ALS detected genotypic variability to that reported in the Caucasian population. In this instance, the authors argued against the theory of a single founder effect in C9orf72 repeat expansion.30
Clinical characteristics of classical and atypical phenotypes of ALS and other motor neuron disorders
In a genetic survey of Taiwanese patients with ALS, SOD1 was the most common gene implicated, followed by TARDBP.24 Mutations in FUS also appear to occur at a higher frequency in Chinese and Korean cohorts, compared to Caucasians, with ALS.31 ,32 A similar pattern is also seen with OPTN mutations.33 ,34 Although there is heterogeneity in disease-causing genes in ALS from the different populations, the clinical phenotypes of the mutations do not tend to vary.
The advent of genome-wide association studies in Asian populations uncovered two novel loci associated with ALS in a large cohort of Han Chinese patients with ALS (n=1212).35 The two SNPs reside in chromosomes 1q32 and 22p11, respectively. Potential candidate genes in these regions include CMAK1G in chromosome 1, involved in neuronal development, maintenance and function, and CABIN1 and SUSD2 in chromosome 22. SUSD2 is expressed in the brain as well as in the kidneys and lungs, known to induce apoptosis, and while its role remains unknown in motor neurons, it could potentially be involved in neuronal apoptosis, leading to motor neuron degeneration.
In Japanese patients with ALS, a large-scale genetic association study of patients with ALS (n=1305) identified a functional SNP located in an enhancer region of ZNF512B at chromosome 20q13.36 Separately in the spinal cord of patients with ALS, there was greater expression of ZNF512B in the anterior horn cells when compared with controls. ZNF512B is a likely key positive regulator of transforming growth factor beta (TGF-β) signalling that is critical for survival of motor neurons. Decreased expression of ZNF512B would thus lead to decreased TGF-β signalling resulting in increased disease susceptibility. To date, this SNP has also been significantly associated with Han Chinese patients with ALS.37 In a separate study of 465 Japanese patients, linkage disequilibrium block was associated with decreased expression of TTN and rapid functional decline of patients with sporadic ALS.38
In a recent Korean study, multigene panel testing of 18 ALS-related genes followed by exome sequencing was performed in 4 patients with familial ALS and 148 patients with sporadic ALS. Four known mutations in four genes (SOD1, ALS2, MAPT and SQSTM1) and 28 non-synonymous heterozygous variants in nine genes were found in patients with ALS. The findings suggested targeted multigene panel testing as an efficient method of screening for ALS-related genetic mutations.39
In India, an L84F mutation in exon 4 of SOD1 with a novel nucleotide variation, c.255G>T was identified in four members of a family with autosomal-dominant inherited ALS. The mutation was not detected in 48 patients with sporadic ALS or 35 age-matched healthy controls. Of interest, patients with ALS with the mutation demonstrated phenotypic variability in terms of age of onset, site of disease onset and duration of survival.40
Atypical Asian phenotypes
Aside from the conventional presentations of ALS, there are several atypical presentations of motor neuron disorders or motor neuronopathies that appear to have a predilection for Asian patients (table 3). It is less clear if these conditions form part of the ALS spectrum, representing a less aggressive form of disease in some, or if they are completely different diseases with different risk factors.
Juvenile muscular atrophy of distal upper extremity
The initial descriptions of monomelic atrophy date back to 1959 by Hirayama et al41 who described juvenile muscular atrophy of the unilateral upper extremity in Japanese patients. The condition continues to be referred to by its eponymous name, Hirayama disease, although Hirayama has since advocated use of the more descriptive term, juvenile muscular atrophy of distal upper extremity.42 In 1984, Gourie-Devi et al43 described similar patterns of weakness in patients from India, referring to the disease as monomelic amyotrophy to reflect the involvement of the lower as well as upper limbs. Although there have been descriptions of such cases in the western population, there is a disproportionately greater number of patients of Asian descent who are afflicted.
In juvenile muscular atrophy of distal upper extremity, the diagnosis is made on the basis of distally dominant muscle weakness with age of onset from the teenage years up until early adulthood. There appears to be a preponderance of males accounting for almost 90% in one large series from Japan.44 An initial period of progression over a few years is typically followed by a plateau. The pattern of muscle atrophy affects the hand muscles and the ulnar aspect of the distal forearm or ‘oblique amyotrophy’, usually sparing the brachioradialis muscle (figure 2A). There is marked weakness with cold exposure referred to as ‘cold paresis’ and hand tremor. Electromyography shows neurogenic changes, and in >70% of cases, these changes extend to the unaffected upper limb.

{kind=link}
{kind=link}
Clinical and imaging features of juvenile muscular atrophy of distal upper extremity (Hirayama disease). (A–B) Typical pattern of intrinsic hand muscle atrophy and wasting of forearm muscles with brachioradialis relatively well preserved. The C7-8-innervated muscles are preferentially affected giving rise to ‘oblique amyotrophy’, with the border of the atrophy passing obliquely from the mid-radial region to the elbow. There is debate as to the pathophysiology of this disproportionate wasting. One theory is forward displacement of the posterior dural wall with neck flexion due to an imbalance in the development of spinal cord and spinal column. Forward neck flexion from neutral (B) results in lower cervical cord flattening and forward shift of the posterior dural wall (C).
Cervical cord imaging in these patients has typically identified forward displacement of the cord, with asymmetrical cord atrophy. In the forward neck flexion position, flattening of the lower cervical cord and forward displacement of the posterior dural wall have been demonstrated on imaging45 (figure 2B, C). These findings led to the hypothesis that the disease entity is the result of imbalance between the development of the spinal cord and spinal column, resulting in a form of myelopathy with sustained neck flexion. Measures to immobilise the neck with collars have had modest effects.46 ,47 Similar findings have also been found in other patient series.48 ,49 Although the hypothesis offers an attractive perspective, it does not explain why patients without the aforementioned MRI changes have the disease. It also does not explain why this entity is seen predominantly in Asian patients. The latter suggests that other factors that are inherent to this population increase their susceptibility to developing the condition.
Monomelic amyotrophy is a term that has been synonymously used with juvenile muscular atrophy of distal upper extremity as both conditions share many clinical features.43 ,50 Subsequently, Nalini et al51 reported their findings in 279 patients of Indian descent over a 35-year period. Monomelic amyotrophy represented 11.3% of the overall disorders of the motor neuron with ALS accounting for 83% of cases. Almost 20% of their patients with monomelic amyotrophy had atrophy of one lower limb. Male predominance was evident with ratios of 9:1 in upper limb atrophy and 13:1 in lower limb atrophy. Interestingly, cold paresis was only reported in patients with upper limb atrophy. The cases were sporadic in onset suggesting that a forme fruste of spinal muscular atrophy was not likely.
In 20 cases where MRI in neck flexion was performed, forward displacement of the cervical cord was seen only in eight patients. Similar to juvenile muscular atrophy of distal upper extremity, the prognosis in monomelic amyotrophy, including those with lower limb involvement, is good. Long-term follow-up studies over a period of nearly 10 years showed that in 95%, there was no progression beyond 5 years and no patients developed the features of ALS.52
Madras motor neuron disease
In 1970, an unusual pattern of ALS was described to occur in young patients from Madras (now known as Chennai) in South India.53 Patients typically had a thin body habitus and presented with muscle wasting and weakness in the distal limb, facial and bulbar muscles. Associated with this are pyramidal signs and sensorineural deafness. In some cases, optic atrophy, cerebellar involvement and chin fasciculations were also present.54 ,55 There have been single case reports from other countries including China, Thailand and Turkey.
In a large series of 116 patients from India, the majority of patients (98%) were from various parts of South India.56 The mean age of onset was 15.8 years, affected males and females equally and almost 77% of cases were sporadic. Involvement of the lower cranial nerves and pyramidal tract signs distinguished the condition from monomelic amyotrophy, likening it instead to classical ALS. However, the presence of sensorineural deafness and optic atrophy would be unusual and raises the possibility of mitochondrial dysfunction. Complete mitochondrial DNA sequencing of 45 patients and their maternal relatives revealed 396 variations, although it remains unclear if any of the variants were pathogenic.57
The underlying pathophysiology of Madras motor neuron disease is still unknown, although the clinical features suggest that it goes beyond the anterior horn cells. In one autopsy study, there was severe loss of anterior horn cells, along with demyelination and sclerosis of the ventrolateral columns. Changes in neuronal depletion and marked gliosis were seen in the cochlear nucleus with demyelination and axonal loss of the cochlear nerve. The findings were suggestive of an inflammatory process.58 In support of this, a case report of a trial of intravenous immunoglobulin in one young Chinese patient with Madras motor neuron disease was associated with an improvement.59
ALS-parkinsonism-dementia complex
In the mid-1940s, the first report emerged of a high incidence of a fatal form of ALS, unusually associated with parkinsonism and dementia in Guam, which was followed by similar reports in other areas of the Western Pacific. The areas affected were Chamarros, Guam,60 the Kii Peninsula in Honshu Island, Japan61 and the southern West Papua, Indonesia.62
The clinical features of ALS are similar to those seen in classical ALS with upper and lower motor neuron involvement. However, patients also displayed parkinsonian features of bradykinesia, rigidity and tremor. Neuropsychological assessment showed marked abulia, apathy, bradyphrenia and hallucinations.63 Any of the three clinical entities can be the initial presenting feature. Pathological studies showed neurofibrillary tangles in the cortex and brainstem of patients with parkinsonism-dementia complex and, to a lesser extent, ALS. The spinal cord showed marked degeneration and atrophy of the corticospinal tract and loss of anterior horn cells.64
Although there are sporadic cases, the majority of patients describe a family history. In a single family, the phenotype can range from just ALS, Parkinson's disease or dementia to a combination of all three.65 A causative gene has yet to be identified and it is likely that the disease is not one of straightforward Mendelian inheritance. Of the many postulations, environmental factors are thought to play an important role in disease development. A recurring hypothesis is chronic exposure to the neurotoxin, β-N-methylamino-l-alanine, which is present in cycad seeds.66
Over time, there has been a marked decline in the incidence, particularly that of ALS, in the region.67 In the Kii peninsula, the incidence of ALS remains higher than that in other Japanese regions. In one study, the authors found the C9orf72 repeat expansion in 3/15 (20%) patients from the southernmost region of the Kii peninsula.68 Family history was present in only one of the three cases. This frequency was in sharp contrast to 2.5% (1/40) in familial and none in patients with sporadic ALS from other regions in Japan. Their findings suggest that the C9orf72 repeat expansion may partially account for the higher frequencies in the Kii peninsula.
Disease progression and survival in Asia
Reports of classical ALS phenotypes in the Asian population suggest similar subtypes to those seen in the Caucasian patients, with limb-onset ALS accounting for the majority (around 75%) followed by bulbar-onset ALS.13 ,14 ,16 ,69 Compared with the mean disease survival in ALS from western populations, a recent study of 549 Japanese patients with sporadic ALS reported a longer median survival (without the use of tracheostomy positive pressure ventilation (TPPV)) at 48 months.11 From that cohort, older age of onset, initial presentation of neck weakness, proximal dominant muscle weakness in the upper extremities and non-use of riluzole were associated with a reduced survival outcome. In the study, 58.8% of patients were treated with riluzole. Significantly, this study further highlighted findings from a previous study in the Japanese cohort which identified neck weakness as an independent prognostic factor for survival and functional deterioration.70
In Taiwan, median survival was 66.6 months (including those with tracheostomy) and poor prognostic factors included rural dwelling and lower socioeconomic status.71 Patients on riluzole and with tracheostomy had the highest rate of survival. Despite its known modest benefit, 40% of patients refused riluzole and a further 43% stopped taking the medication. The median survival in Chinese and Indian studies suggests longer periods of survival in association with an earlier age of onset.13 ,14 ,16 Migration studies have also observed similar patterns. In one study of migrants from the Indian subcontinent to the UK, a younger age of onset of patients with ALS with lower mortality rates was also observed.72
A significant difference in the management of patients with ALS in Asia is the high administration of TPPV in countries like Japan, South Korea and Taiwan. The utility of TPPV ranges from 25% to 46% in Japan73 and is 21% in Taiwan.71 This is in sharp contrast to the West where the use of TPPV is reported to be fewer than 10% of patients with ALS. 73 In a study that investigated the role of Japanese and American neurologists in patients' choice for TPPV, several differences were apparent.73 Although both share similar views on the management of ALS and their own personal preference against the use of TPPV, differences in national health policies contributed to variations in individual practice. In the USA, patients with ALS were treated in large specialised centres where palliative care was provided and end-of-life and quality-of-life issues were addressed early on. In Japan, use of TPPV is advocated by clinicians and patient organisations and withdrawal of ventilation receives less support from the institution. In patients on prolonged TPPV of >9 years, ophthalmoplegia was observed in 30% of patients, occurring at a higher frequency in patients with an earlier age of onset.74
Use of enteral tube feeding through either percutaneous gastrostomy or nasogastric tubes in ALS differs between the Asian countries. In Japan, 36.5% of patients have a gastrostomy tube in place.74 The frequency is lower in South Korea and Taiwan, respectively.12 ,71 In China, 56% of patients with ALS (n=441) presenting to urban tertiary centres were on either gastrostomy or nasogastric feeding.75 This was in sharp contrast to a different study of patients with ALS from the less-developed South-West China where only 15 of 1116 (0.01%) patients were reported to have percutaneous gastrostomy.14
One of the biggest challenges in the management of ALS across Asia relates to the implementation of evidence-based treatment. In countries where the government absorbs the cost of caring for patients with ALS, such as Japan, South Korea and Taiwan, life-prolonging measures such as riluzole, ventilatory support and artificial feeding are provided to patients. Similar access to such care may not be available to patients from countries where financial resources are limited and the healthcare costs are borne by patients. This has been shown at least in studies from India, Malaysia and China where limitations exist on patients' affordability of treatments as well as patients' willingness to accept life-prolonging measures such as artificial feeding and respiratory support.13 ,16 ,69 There are circumstances where the choice of not accepting life-prolonging interventions relates to cultural factors, but it is likely that financial costs are prohibitive. It is therefore interesting to note that the median survival in China and India are reportedly higher than those in the West. This raises the possibility that these groups of patients may harbour an inherent protective factor which allows for longer survival.
Non-motor features of ALS
It is widely accepted that ALS is a heterogeneous disease and patients can exhibit non-motor features such as cognitive impairment and behavioural changes. Studies from the West have cited that between 20% and 50% of patients with ALS would fulfil a probable or definite diagnosis of FTD.76 These psychological changes are important to recognise in patients with ALS as the presence of dementia and apathy has an adverse effect on patient care, patient autonomy and overall patient survival.
Very few Asian studies have investigated the cognitive and behavioural components of ALS. The frequencies with which they occur in Asian patients with ALS are less clear. In a cohort of 166 South Korean patients with sporadic ALS, 42% of patients were found to have behavioural or cognitive dysfunction.77 None carried the C9orf72 repeat expansion, which accounts for the highest proportion of ALS/FTD overlap in the West, suggesting that a different genetic mutation may be responsible.5 Patients with cognitive impairment had reduced survival. In one imaging study of 21 Japanese patients with ALS, apathy was demonstrated during the early course of the illness and its severity was significantly associated with frontal lobe atrophy, lending further support that a continuum exists between ALS and FTD.78 Further evidence for this was demonstrated in the pathological study of Japanese patients with FTD, FTD/ALS and ALS/FTD.79 Pathological changes of TDP-43 were seen in the anterior horn cells of patients across the spectrum, with the most severe neuronal loss and gliosis in patients with ALS/FTD. Shorter survival times were also found in patients with ALS with FTD overlap. However, in a different study of 126 sporadic Chinese patients, the authors found that frontal lobe dysfunction did not affect the disease progression or survival.80
Future considerations for the Asian region
Asia represents a diverse population and there is evidence to suggest that populations arising from mixed ancestry have reduced susceptibility to developing ALS.6 Population-based studies, which are currently lacking in Asian cohorts, will be increasingly important to accurately define the true prevalence and incidence of ALS in Asia. Our current understanding also suggests heterogeneity in the clinical presentation and genetic basis of ALS in Asia. There is evidence to suggest that Chinese and Indian patients with ALS have younger age of disease onset but a longer median survival, despite not receiving disease-modifying therapy such as riluzole or ventilatory support, raising the possibility of inherent protection. The recently discovered C9orf72 repeat expansion accounts for <2% of familial or sporadic ALS in Asia, which is in sharp contrast to the West, where the mutation represents 40% of familial ALS and up to 20% of sporadic ALS. Genome-wide association studies have also identified potential disease-causing SNPs unique to Asian patients with ALS. Asia represents >50% of the world population and recent migratory patterns have observed a considerable number of people from Asia migrating to the West. In such instances, the atypical patterns of ALS that are almost exclusively seen in patients of Asian descent have prevailed, suggesting that genetic predisposition rather than environmental influences are at play. However, to date, the genetic causes of these conditions remain undiscovered. This may be due to the reduced power of genetic association studies, which could be overcome through collaborative efforts where a larger and diverse population can be studied.
Currently, there have been numerous multicentre clinical trials in ALS, but few have incorporated the Asian patient cohort. The recent phase III EMPOWER study investigated the role of dexpramipexole in ALS, following the positive findings that were observed in the phase II study.81 Unfortunately, despite being well tolerated, dexpramipexole showed no significant differences in disease end points of ALS and controls. However, the study was performed exclusively in countries with a Caucasian majority, thus introducing ethnic and likely genetic bias. Similarly, there have been several clinical trials that have been performed exclusively in Japanese or Korean cohorts. These include ursodeoxycholic acid, mexiletine, edaravone and high-dose methylcobalamin.82–85 Future clinical trials are best served by being more inclusive, incorporating patient cohorts with ALS from the different regional registries. Subgroup analyses could then be performed to investigate the performance of the study drug in patients from different ethnic backgrounds.
The apparent discrepancies between Asian and Western cohorts with ALS merit further study to explore the differences in heterogeneity between the two cohorts. In an effort to achieve similar standards of care, treatment and research in the region, an Asia-Pacific working group of ALS clinicians and researchers was established in 2014, referred to as the Pan-Asian Consortium for Treatment and Research in ALS (PACTALS). The Consortium aims to provide a platform in which member countries are able to form effective collaborations and participate in activities within the greater international ALS community (http://pactals.org/).
Few medical advances are made in isolation and new solutions require collaboration across our regions. The synergies achieved by establishing an active collaboration of clinicians and researchers from the basic, translational and clinical neurosciences in Asia-Pacific and the rest of the world will promote new research discoveries into ALS, providing hope for those individuals and families whose lives are devastated by this condition.
Acknowledgments
NS is a recipient of the 2015 Australian Endeavour Executive Fellowship.
References
Footnotes
Contributors NS contributed to the conception and initial draft of the manuscript and figures. Table 3 was prepared by KS and the initial draft of figure 1 was prepared by YN. All the coauthors contributed to the editing of the manuscript, with final editing being undertaken by NS and MCK.
Patient consent Obtained.
Provenance and peer review Commissioned; externally peer reviewed.