Article Text

Abstract
Background The approval of 9-δ-tetrahydocannabinol and cannabidiol (THC:CBD) oromucosal spray (Sativex) for the management of treatment-resistant multiple sclerosis (MS) spasticity opened a new opportunity for many patients. The aim of our study was to describe Sativex effectiveness and adverse events profile in a large population of Italian patients with MS in the daily practice setting.
Methods We collected data of all patients starting Sativex between January 2014 and February 2015 from the mandatory Italian medicines agency (AIFA) e-registry. Spasticity assessment by the 0–10 numerical rating scale (NRS) scale is available at baseline, after 1 month of treatment (trial period), and at 3 and 6 months.
Results A total of 1615 patients were recruited from 30 MS centres across Italy. After one treatment month (trial period), we found 70.5% of patients reaching a ≥20% improvement (initial response, IR) and 28.2% who had already reached a ≥30% improvement (clinically relevant response, CRR), with a mean NRS score reduction of 22.6% (from 7.5 to 5.8). After a multivariate analysis, we found an increased probability to reach IR at the first month among patients with primary and secondary progressive MS, (n=1169, OR 1.4 95% CI 1.04 to 1.9, p=0.025) and among patients with >8 NRS score at baseline (OR 1.8 95% CI 1.3–2.4 p<0.001). During the 6 months observation period, 631(39.5%) patients discontinued treatment. The main reasons for discontinuation were lack of effectiveness (n=375, 26.2%) and/or adverse events (n=268, 18.7%).
Conclusions Sativex can be a useful and safe option for patients with MS with moderate to severe spasticity resistant to common antispastic drugs.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Spasticity, defined as abnormally increased muscular tone, is a common symptom in patients with multiple sclerosis (MS) patients.1 MS spasticity causes or worsens different MS symptoms, usually having a big impact on patients' quality of life.2 The most frequent symptoms associated with spasticity in patients with MS, beyond mobility worsening, are spasms, pain, poor sleep quality (linked to pain and nocturnal spasms) and urinary dysfunction.3 Classically recommended medications for generalised spasticity, including baclofen, tizanidine, dantrolene, benzodiazepines and clonazepam, have shown limited clinical benefit in systematic reviews.4 ,5 The approval of 9-δ-tetrahydocannabinol and cannabidiol (THC:CBD) oromucosal spray (Sativex), in a number of European countries, including Italy, provides a new opportunity as an add-on medication for the management of moderate to severe generalised spasticity and related symptoms in patients with MS resistant to common oral antispastic drugs.6 ,7 Sativex is an endocannabinoid system modulator containing THC and CBD in a near 1:1 ratio. THC interacts with cannabinoid human receptors that play a key role in the modulation of muscle tone, whereas CBD at higher than natural concentrations may limit the psychoactive effects of THC.8 The efficacy of Sativex oromucosal spray as an add-on therapy for symptoms improvement in patients with MS with moderate to severe MS spasticity has been demonstrated in several clinical trials.6 ,9–12 The largest pivotal phase III, enriched-design clinical trial included patients with MS with moderate–severe spasticity corresponding to a patient-reported numerical rating scale (NRS) score ≥4.
In this clinical trial, after a first 4 weeks single-blind THC:CBD trial period, 47% of patients showed an initial improvement in spasticity evaluated by the MS spasticity 0–10 NRS scale (defined as a ≥20% reduction vs baseline). During the second phase, consisting of a double-blinded randomisation of the initial responders to continue either with THC:CBD treatment or placebo, a significantly greater proportion of patients receiving THC:CBD achieved a clinically relevant response (defined as a ≥30% reduction in their baseline NRS score). Almost 47% of patients reported adverse events (AEs), with mild to moderate dizziness and fatigue being the most common treatment-related adverse event.12 Although clinical trials are needed to evaluate the efficacy and safety of a new compound to defend its approval, observational studies are essential to depict the effect of treatment in real-world conditions.13 Postapproval surveillance registries have been established to evaluate possible short-term and long-term risks associated with Sativex use.14 ,15 The MOVE 2 German observational study (n>300) of THC:CBD oromucosal spray showed results aligned with those from clinical trials, with 42% of patients being initial responders after 4 weeks.16 Another recent prospective observational study carried out in Spain (n>200) provided data about 2/3 of patients still on prescription after 6 months from treatment start.17 In these observational studies, patients tended to use lower doses compared with clinical trials (6–7 sprays/day vs >8) and the incidence of AEs was lower, with no evidence of addiction, abuse, misuse or memory impairment.17 Given the growing importance of data derived from real-world observational studies, we decided to collect information about Sativex use in Italy, using the Italian medicine agency (AIFA) prospective e-registry, mandatory for all patients receiving a Sativex prescription in Italy, as a main source document, considering that, after the large sample was collected, much larger than that in the available THC:CBD studies so far, it would bring a comprehensive picture of its effect, minimising biases. For further information, we also used complementarily the involved patients' medical charts retrospective review.
The main aim of our study was to describe Sativex effectiveness and AEs, including possible evidence of abuse or misuse in a large population of Italian patients with MS.
Materials and methods
Design and setting
This was a multicentre observational study aimed at collecting prospective reported data to evaluate Sativex effects (effectiveness and tolerability) in a large population of Italian patients with MS with resistant spasticity carried out in a routine outpatient setting. All patients included in the study were treated in accordance with the approved label and expected standards of good clinical practice. Complementary clinical and demographic parameters were acquired retrospectively from the patients' medical records.
The study was approved by the Policlinico-Vittorio Emanuele (Catania, Italy) Ethics Committee (number 37/2015/PO) and by the Ethics Committee of the other participating centres.
Patients were consecutively included in the study at the start of Sativex treatment (baseline) and followed up over 6 months, with data collection at baseline, after 4 weeks (T1), after 12 weeks (3 months, T2) and after 24 weeks (6 months, T3) from baseline.
Patient population
The AIFA registry requests that patients are eligible for starting Sativex treatment when fulfilling the following approved label-related inclusion criteria: patients with MS older than 18 years, with moderate to severe spasticity (0–10 NRS score ≥4) and not responding to common and ongoing antispastic drugs (used under their approved label). Exclusion criteria are: severe cardiovascular diseases, history of psychiatric diseases, use of street cannabis and/or other psychoactive drugs and/or MS spasticity NRS score <4.
In our study, we collected data from all the patients starting Sativex therapy between 1 January 2014 and end of February 2015 in the participating centres.
Data collection
We collected data from the AIFA Sativex e-registry website. The MS spasticity evolution was evaluated by the validated 0–10 NRS patient-rated scale (0=no, 10=maximal spasticity).18 MS physical disability was evaluated using the expanding disability status scale (EDSS). Other parameters such as use of other antispastic drug, previous antispastic drugs and treatment discontinuation were collected.
Furthermore, complementary demographical and clinical history data, tolerability, daily dose (number of puffs per day), clinical response to Sativex, discontinuation reasons and time to discontinuation, and improvement of spasticity-associated symptoms (stiffness, spasms/cramps, clonic movements, sleep disturbances, urinary dysfunctions, pain, depressed mood) were collected from patients' medical charts and using an ad hoc interview. Data were manually entered in an ad hoc created database. The following definitions were used for classifying responses: Sativex effectiveness was evaluated through the initial response (IR) threshold, defined as ≥20% NRS spasticity score improvement versus the baseline value,18 and clinically relevant response (CRR) threshold, defined as ≥30% NRS spasticity score improvement versus baseline value.18 Tolerability was assessed collecting each AE and serious adverse event (SAE) occurring during the whole study period, in accordance with the pharmacovigilance regulations.
Statistical analysis
Data were analysed using the STATA V.11.0 software packages (StataCorp. 2011. Stata Statistical Software: Release 12. College Station TSL). Data cleaning was performed before the data analysis considering both range and consistence checks.
Quantitative variables were described using means and SDs. The difference between means and the difference between proportions was evaluated by the t test and the χ2 test, respectively. A Shapiro-Wilk test was performed to assess the normal distribution of data. In case of a not normal distribution, appropriate non-parametric tests were performed. Unconditional logistic regression analysis was performed, and for each study variable we calculated ORs, 95% CIs and p values (two-tailed p≤0.05 significant level). Univariate logistic regressions were fit considering binomial ‘IR yes/no’ as a dependent variable and age, sex, disease duration, MS type, baseline EDSS and baseline NRS as independent variables. Whenever quantitative variables were dichotomised, the cut-offs were derived using the median. Multivariate analysis was performed to investigate the independent effect of a risk or protective factor after adjustment for one or several other factors or to adjust for confounding variables. Parameters associated with the outcome in the univariate analysis with a threshold of p=0.10 were included in the multivariate model.
Results
Patient population
A total of 1615 patients with MS spasticity starting treatment with Sativex were recruited from 30 large Italian MS centres distributed geographically across the nation from about 160 specialised MS all-size centres in the country. Recruited patients started treatment between 1 January 2014 and end of February 2015 (see table 1 for demographic and clinical details). Of the 1615 patients, 18 were excluded from the analyses because baseline NRS was not available. A total of 1432 patients (89.7% from baseline) reached the end of the first month trial period T1 and were eligible for the trial period early response effectiveness analysis. One hundred and sixty-five patients had to be excluded for different reasons (see figure 1). At T2 (3 months), 889 (55.6%) patients and at T3 (6 months), 593 patients (37.1%) were included in the analysis (see figure 1). Reasons for not including the remaining patients at T2 and T3 are detailed in figure 1. During the whole observation period, a total of 631 patients (39.5%) discontinued therapy.
Clinical and demographic data

Patient flow chart. *Data not available at the specific time point.
Effectiveness outcome
The mean±SD 0–10 MS spasticity NRS score value at baseline for the analysable 1597 patients was 7.5±1.4.
A statistically significant difference was found comparing the baseline score with the different time points NRS score values of the remaining patients. At T1, the NRS score was 5.9±1.6 (p<0.0001, n=1432), at T2 5.1±1.6 (p<0.0001, n=889) and at T3 4.8±1.7 (p<0.0001, n=593; see table 1).
Within the 1432 patients who reached T1, end of trial period, 4 weeks after treatment start, a total of 937 patients (65.4%) were considered clinical responders according to the treating specialist's overall clinical judgement. One hundred and thirty-three patients (9.3%) were considered partial responders (suitable to become a responder in longer follow-up), whereas 349 patients (24.4%) were deemed to be non-responders. No data were available for 16 patients (1.1%).
Regarding the NRS scale measurement of the MS spasticity evolution, at T1 1010 patients (70.5%) reached a ≥20% NRS score reduction compared with baseline (IR threshold), and 405 (28.3%) had already reached a≥30% NRS reduction (CRR). We found a 22.6% reduction of mean NRS score at T1 compared with baseline (n=1432). Within the T1 IR patients subgroup, we found a 30.3% NRS score mean reduction at T1; and considering T1 CRR patients, we found a 40.8% NRS score mean reduction at T1 (figure 2).

Multiple sclerosis spasticity NRS evolution between T0 and T1. NRS, numerical rating scale.
A total of 889 patients reached the 3 months' visit T2, which is 84.4% of those who might have reached it on the basis of the treatment start date, n=1053. They showed a mean NRS score of 7.5±1.5 at baseline and 5.1±1.6 at T2 (32% mean NRS reduction). A total of 779 patients maintained scores ≥the IR threshold at T2 (88% of patients reaching this visit and 73.9% of those who should have reached it), with a mean NRS reduction of 31.6% at T2 compared with baseline (7.6±1.5 vs 5.2±1.4). Three-hundred and eleven patients reached the CRR threshold at T2 (35% of those reaching the visit and 29.5% of those who should have reached it) with a mean NRS reduction of 41.5% versus baseline (7.7±1.5 vs 4.5±1.4).
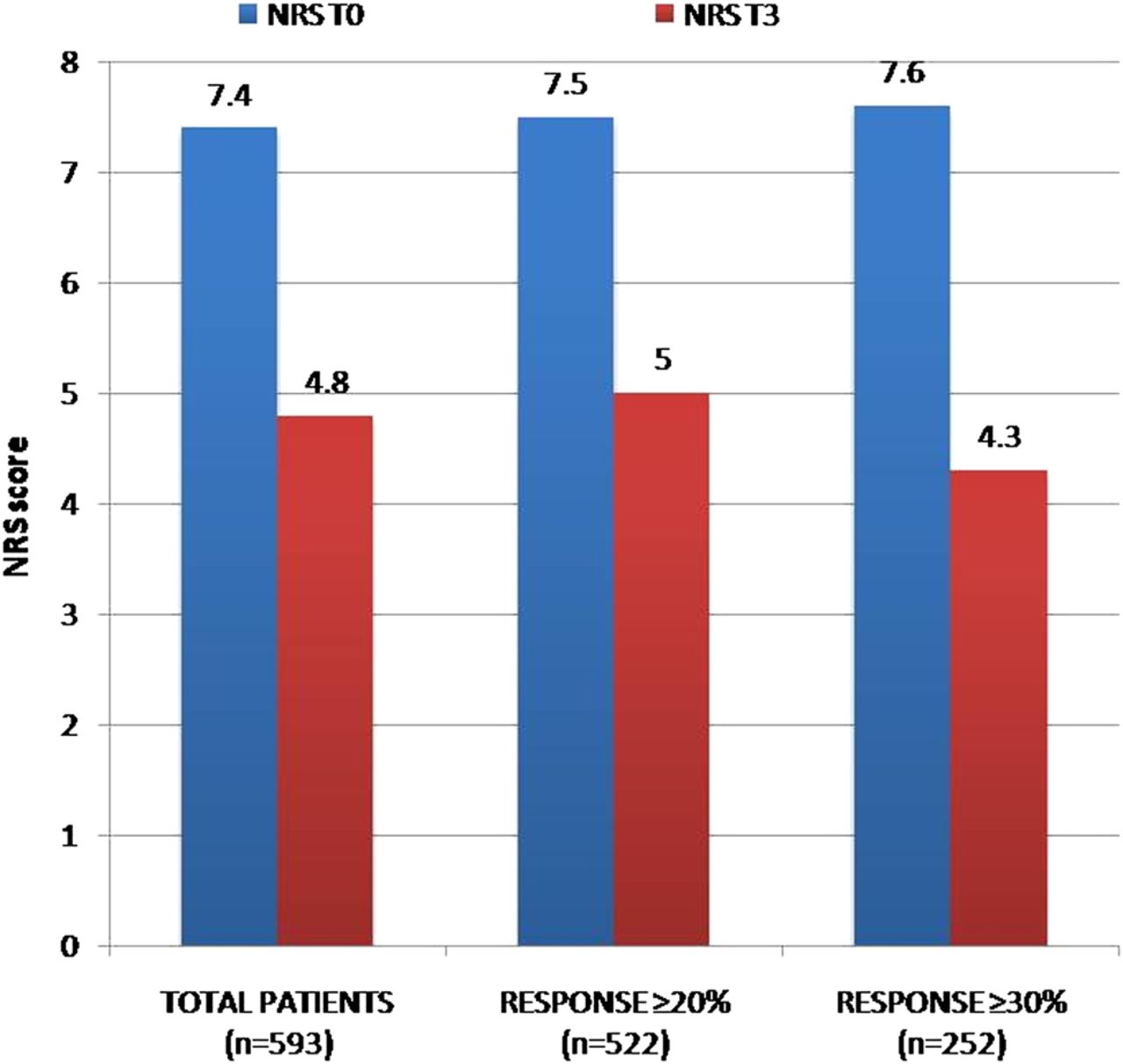
A total of 593 patients were available for analysis at the T3 sixth months' control visit (73% of those who might have reached it according to start date, n=811). Their mean±SD MS spasticity 0–10 NRS score at T0 was 7.4±1.5, and we found a reduction of 35% at T3 compared with baseline (T3 NRS mean score 4.8±1.7). Five hundred and twenty-two maintained at least a ≥IR threshold score at T3 (88% of patients reaching this visit and 64.4% of those who could have reached it) with a mean NRS reduction of 33.3% (7.5±1.5 at T0 vs 5±1.5 at T3). Considering the CRR population (n=252, 42.5% of patients reaching this visit and 31% of those who could have reached it), we found a mean NRS reduction of 43.4% (7.6±1.5 vs 4.3±1.5; see figure 3).

{kind=link}
{kind=link}
{kind=link}
Multiple sclerosis spasticity NRS evolution between T0 and T3. NRS, numerical rating scale.
Regarding the medication dose, the mean number of puffs per day was 6.8±2.6 at T1 and decreased to 6.5±2.6 at T2 (p=0.007) and 6.3±2.8 at T3 (p=0.0001; see table 1).
Univariate analysis logistic regression showed a significant association between baseline NRS score and MS type and the probability to reach IR at T1 (see table 2).
Univariate analysis of clinical and demographic predictors of NRS IR at T1 in a population of 1432 patients
At a multivariate analysis, an age-adjusted and sex-adjusted significant association with MS course and baseline NRS was found. In particular, according to the multivariate analysis, we found an increased probability to reach IR at T1 among patients with secondary progressive primary progressive (SP-PP) (progressive) MS (n=1169, OR 1.4 95% CI 1.05 to 1.86, p=0.018) and among patients with higher spasticity NRS score (>8) at baseline (n=358, OR 1.8 95% CI 1.4 to 2.4, p<0.001; see table 2).
Reasons for discontinuation
The number of patients who discontinued therapy during the observation period was 631 (39.5%). Of them, 396 patients (24.8% from the overall sample) did not reach the IR threshold. Three hundred and seventy-four patients (23.4% of the overall sample) discontinued after the 4-week trial period. Reasons for discontinuation during the whole observation period were (multiple answers possible) lack of effectiveness (n=371, 23.2%, mostly early), AEs (n=260, 16.3%), non-adherence (n=12, 0.8%), loss at follow-up (n=7, 0.4%) patient's choice (n=5, 0.3%) or reasons not available (n=32, 2%).
Tolerability and safety
AE leading to discontinuation (18.7% of patients reaching T1, 1432 patients) were basically cognitive/psychiatric disturbances (n=55, 3.9% of patients), in particular, out of the 55 cognitive/psychiatric AEs, 9 were cognitive (3 attention deficit, 3 memory impairment, 3 cognitive worsening) and 46 were psychiatric (32 confusional state, 9 panic attack/anxiety disorder, 3 hallucinations, 1 depressive syndrome, 1 suicidal thoughts). Fatigue (n=36, 2.5%), drowsiness (n=32, 2.2%), dizziness (n=30, 2%), gastrointestinal symptoms (n=21, 1.4%), mouth discomfort (n=10, 0.7%), allergic reaction (n=3, 0.2%) and other neurological symptoms (n=16, 1.1%) were other reported AEs, all considered drug-related by clinicians. Data were not available for n=100 (7%). No abuse, addiction or misuse hints were detected. These AEs were considered drug-related. Of 268 patients, 260 discontinued treatment after the onset of AE (see table 3). All patients recovered after discontinuation, the remaining eight being kept on Sativex treatment. We found five patients (0.3% of patients) presenting with a SAE: a hypertensive crisis, one death after acute myocardial infarction, an acute renal failure in a patient with chronic kidney disease, a laryngeal carcinoma case and a breast cancer case. All these SAEs were deemed to be unrelated to the study drug.
AE leading to discontinuation in the population reaching T2 (n=1432)
Discussion
In this large multicentre study, we found that 70.5% of patients reached the IR threshold and that 28.2% had already reached the CRR threshold at the end of the 4-week trial period. According to the physician's judgement, 65.4% of patients were considered responders after the first month of treatment, aligning with the proportion of patients reaching IR (70.5%), whereas 9.3% were considered partial responders, suitable to become responders in a longer follow-up.
We also found in patients reaching T1 (n=1432) a 22.6% mean NRS spasticity score reduction at T1 (first month) and a 35% mean NRS score reduction at T3 (6 months) compared with baseline, showing a decrease in NRS score from 7.5 at baseline to 5.8 at T1 and 4.8 at T3. Considering the IR population, we found a mean 30.3% reduction at T1 and a 33.3% reduction at T3. Considering the CRR group, we found a 40.8% reduction at T1 and 43.4% reduction at T3. These data suggest that a meaningful MS spasticity NRS reduction is obtained during the first month and that it is maintained or slightly increased over time (see the T3 6-month follow-up data). This is in line with previous observational studies.16 ,19 ,20 It is noteworthy that the mean number of puffs decreased slightly from 6.8 at T1 to 6.3 at T3, suggesting that effectiveness may be maintained with lower doses compared to clinical trials (mean number of 8.1 puffs/die in clinical trials),12 while ruling out the need to increase dose to maintain effect over time (tolerance) and even abuse/misuse trends.
A certainly higher proportion of patients in our study reached the IR ≥20% threshold in the first month (70.4%) when compared with patients from the German MOVE 2 study (41.7%). However, in the German study, clinicians reported observing clinical relief of resistant spasticity in 74.6% of patients in the first month of treatment according to their own judgement, in line with the results of our study (70.4%).16 In Italy The AIFA e-registry requires an IR threshold attained after the 4- week trial period to continue with the THC:CBD prescription, obtained through an automatic calculation and within a strict time frame. In other countries (Germany included), the NRS improvement is calculated by the clinician without the use of a compulsory web-based registry, thus possibly affecting the perceptions of the initial response. This is confirmed by another recent observational Italian study, showing that a proportion of 71.7% reached the IR with a mean NRS score reduction of 32%, in line with our results.20 The German MOVE 2 study IR patients' subgroup showed a mean reduction of NRS score of 40.2% at T1, while in our study a lower mean reduction, 30.3%, was observed. Considering the CRR ≥30% spasticity NRS change versus baseline threshold patients subgroup evolution, a somehow larger mean reduction of 49.9% was observed at T1 in Germany,16 while our results showed a mean reduction of 40.8% in this subgroup. In a phase 3 randomised placebo-controlled study, participants were also treated with THC:CBD as an add-on therapy in a single-blind manner for 4 weeks. After 4 weeks, 47.5% of patients reached IR, showing a MS spasticity mean NRS score reduction of 47.6% compared with baseline NRS, higher than the MOVE 2 observational study score of 41.7% but still lower than our 70.4% rate.12 Our results showed a higher proportion of IR responders but a smaller mean change in term of NRS between baseline and T1, not fully in accordance with previous studies, although in line with recent findings from an observational Italian study.20 This could in part be explained by the AIFA web registry features, not allowing an NRS score <4 at baseline and always requiring the IR threshold to be reached at the trial period follow-up visit, thus affecting the recording of the improvement. Indeed, our population seems to be more impaired, in terms of mean EDSS and NRS scores, and has a longer disease duration, when compared with other studies (see table 4). This could in part be explained by multivariate analysis findings showing that reaching the trial period IR threshold could be influenced by a baseline NRS score and disease course (progressive MS). The effectiveness of Sativex on patients with progressive MS spasticity has been investigated by a recent study evaluating clinical and neurophysiological effects of treatment. Although no effect on a neurophysiological measure (H reflex) was reported, the study confirmed the clinical benefit (as per the Modified Ashworth scale) in patients with progressive MS, affected by lower limb spasticity.21
Sativex in a former observational study and clinical trial versus our study: comparison of key parameters
This finding may in part be related to the higher percentage of SP and PP compared to relapsing remitting in our study population (80% PP+SP), suggesting that patients with progressive MS are more prone to have a spasticity not responding to common anti-spastic drugs, requiring Sativex treatment. Moreover, we found that patients with progressive MS are older, and have a longer disease duration and higher EDSS and NRS at baseline, suggesting that these differences might in part have biased our findings (see table 1). As in our cohort, it was recently reported that severe spasticity is more frequently observed in wheelchair users while patients able to walk 300–500 m reported a mild to moderate spasticity.7 On these grounds, it is conceivable that patients with a higher NRS score and those with progressive MS could be more satisfied with any improvement in their condition, while patients with lower EDSS scores and thus better walking ability are more demanding in terms of symptoms improvement. Indeed, we should bear in mind that the use of symptomatic treatment is logically mainly based on patients' reported outcomes, rather than on neurological signs examination. However, replication and further analyses are required to better clarify our findings.
About 40% of our population discontinued treatment, in particular 26.2% discontinued for lack of effectiveness and 18.7% for AEs. The most common AEs reported by patients were cognitive/psychiatric effects, fatigue and drowsiness, in line with the tolerability profile reported in other observational studies.14 ,16 ,19
Although this is the largest study reporting Sativex experience in daily clinical practice, our results may be affected by the observational nature of the study. This could in part be mitigated by the use of the AIFA prospective and mandatory e-registry as a source for spasticity NRS information and main patient features. Indeed, in the Italian e-registry, the evaluations are scheduled at 4 weeks from treatment start and then every 3 months, giving us the opportunity to collect data from every centre at the same time points. Another limitation is the lack of spasticity evaluation through the Ashworth healthcare professional impression categorical scale. Although the use of the Ashworth Scale for the assessment of spasticity could be not valid and reliable, results from the German MOVE 2 Study and a recent clinical trial indicated a small but significant improvement.16
Finally, the follow-up period duration (up to 6 months) may be insufficient to assess Sativex effectiveness, safety and tolerability long-term effects.
Besides the aforementioned limitations, this study is the largest multicentre observational study so far available evaluating Sativex effectiveness and tolerability profile in clinical practice. Our results confirm Sativex as an effective and safe option for patients with MS with moderate to severe spasticity resistant to common antispastic drugs. Longer and large multicentre follow-up studies are needed to evaluate treatment effectiveness and tolerability effect in the population with MS. Further exploitation of the AIFA Sativex e-registry data would be desirable.
References
Footnotes
FP and SM contributed equally to the manuscript.
Twitter Follow Ada Francia at @ada.francia@uniroma1.it
Collaborators Benedetti MD, Bertolotto A, Berra E, Bianco A, Buttari F, Cerqua R, Florio C, Fuiani A, Guareschi A, Ippolito D, Nuara A, Palmieri V, Paolicelli D, Petrucci L, Pontecorvo S, Saccà F, Salamone G, Signoriello E, Spinicci G, Russo M, Tavazzi E, Trabucco E, Trotta M, Zaffaroni M.
Contributors FP, SM, CS contributed to the design and drafting for intellectual content of this manuscript. MPA, RB, SB, RBB, VBM, GFC, PC, DC, GC, SC, MD, AF, AG, CG, AG, AI, GL, GTM, MGM, MM, MM, EM, CP, MR, ES, DS, MT, PV, MZ and the SA.FE. study group contributed to the drafting for intellectual content of this manuscript.
Competing interests FP has received honoraria for scientific lectures and travel payment from Biogen, Novartis, Teva, Genzyme, Bayer Schering, Merck Serono and Almirall. SM has received honoraria for scientific lectures from Biogen, Almirall, travel payment from Novartis, Biogen Idec, Genzyme, Almirall Bayer Schering, Merck Serono and Teva, and she serves on the scientific advisory board for Biogen. MPA serves on scientific advisory boards for Biogen and Merck Serono and receives speaker honoraria and research support from Biogen Idec, Merck Serono, Novartis, Almirall and Genzyme. AG has received support for travel to scientific meetings from Almirall, Biogen, Genzyme, Merck-Serono and Novartis. RB has received honoraria for scientific lectures and travel payment from Biogen, Novartis, Teva, Genzyme, Bayer Schering, Merck Serono and Almirall. SB received speaker and advisory board honoraria from Biogen, Novartis and Merck-Serono. RBB received a grant for advisory board activities from Almirall. VBM has received honoraria for scientific lectures and travel payment from Biogen, Novartis, Teva, Genzyme, Bayer Schering, Merck Serono and Almirall. DC is an Advisory Board member of Almirall, Bayer Schering, Biogen, Genzyme, GW Pharmaceuticals, Merck-Serono, Novartis and Teva and received honoraria for speaking or consultation fees from Almirall, Bayer Schering, Biogen Idec, Genzyme, GW Pharmaceuticals, Merck Serono, Novartis, Sanofi-Aventis and Teva. He is also an external expert consultant of the European Medicine Agency (EMA), and the principal investigator in clinical trials for Bayer Schering, Biogen Idec, Merck Serono, Mitsubishi, Novartis, Roche, Sanofi-aventis and Teva. His preclinical and clinical research was supported by grants from Bayer, Biogen, Merck Serono, Novartis e Teva. GC has received honoraria for scientific lectures from Biogen, Novartis, Teva, Genzyme, Bayer Schering, Merck Serono, Almirall. MD has received honoraria for scientific lectures from Novartis, Merck-Serono, Biogen and Teva. AF received a grant for advisory board activities from Almirall. GFC reveived honoraria for speaking activities from Genzyme, Merck-Serono, Biogen and Novartis. CG received honoraria from Biogen, Genzyme, Novartis, Merck Serono, Almirall and Teva. AG received honoraria from Biogen, Genzyme, Novartis, Merck Serono, Almirall and Teva. GL received honoraria from Biogen, Genzyme, Novartis, Merck Serono, Almirall and Teva. GTM received speaking and advisory honoraria from Biogen, Novartis and Teva. MGM has received speaker honoraria and honoraria for serving on advisory board activities from Bayer Schering, Biogen Idec, Merck Serono, Novartis, Sanofi Aventis and Teva, and research grants from Merck Serono and Novartis. MM received honoraria for serving on the scientific advisory boards of Almirall, Novartis, Biogen and Genzyme. MM received honoraria from Biogen, Genzyme, Novartis, Merck Serono, Almirall and Teva. EM received honoraria from Biogen, Genzyme, Novartis, Merck Serono, Almirall and Teva. CP received honoraria for scientific lectures from Biogen, Novartis, Teva, Genzyme, Bayer Schering, Merck Seronoand Almirall. MR received honoraria from Biogen, Genzyme, Novartis, Merck Serono, Almirall and Teva. ES received honoraria from Biogen, Genzyme, Novartis, Merck Serono, Almirall and Teva. MT received honoraria for scientific lectures from Biogen Idec, Novartis, Teva, Genzyme, Bayer Schering, Merck Serono and Almirall. MZ has received compensation for consulting services from Boehringer-Ingelheim, Lundbeck and Union ChimiqueBelge, and scientific grants from AIFA- Agenzia Italiana del Farmaco, Novartis and Lundbeck CS received honoraria from Biogen, Genzyme, Novartis, Merck Serono, Almirall and Teva.
Ethics approval The study was approved by the Policlinico-Vittorio Emanuele (Catania, Italy) Ethics Committee (n° 37/2015/PO) and by the Ethics Committee of the other participating centres.
Provenance and peer review Not commissioned; externally peer reviewed.