Article Text

Abstract
Background TANK-binding kinase 1 (TBK1) gene has been recently identified as a causative gene of amyotrophic lateral sclerosis (ALS).
Methods We sequenced the TBK1 gene in a cohort of 154 Italian patients with ALS with unclear genetic aetiology. We subsequently assessed the pathogenic potential of novel identified TBK1 variants using functional in vitro studies: expression, targeting and activity were evaluated in patient-derived fibroblasts and in cells transfected with mutated-TBK1 plasmids.
Results We identified novel genomic TBK1 variants including two loss-of-function (LoF) (p.Leu59Phefs*16 and c.358+5G>A), two missense (p.Asp118Asn and p.Ile397Thr) and one intronic variant (c.1644–5_1644-2delAATA), in addition to two previously reported pathogenetic missense variants (p.Lys291Glu and p.Arg357Gln). Functional studies in patient-derived fibroblasts revealed that the c.358+5G>A causes aberrant pre-mRNA processing leading TBK1 haploinsufficiency. Biochemical studies in cellular models showed that the truncating variant p.Leu59Phefs*16 abolishes TBK1 protein expression, whereas the p.Asp118Asn variant severely impairs TBK1 phosphorylation activity. Conversely, the p.Ile397Thr variant displayed enhanced phosphorylation activity, whose biological relevance is not clear.
Conclusion The observed frequency of TBK1 LoF variants was 1.3% (2/154), increasing up to 3.2% (5/154) by taking into account also the functional missense variants that we were able to classify as potentially pathogenic, supporting the relevance of TBK1 in the Italian population with ALS.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Amyotrophic lateral sclerosis (ALS) is a severe neurodegenerative motor neuron disease characterised by progressive loss of upper and lower motor neurons leading to death within 2–5 years after diagnosis.1 Cognitive dysfunction occurs in 20%–50% of cases, whereas 5%–15% of patients develop overt frontotemporal dementia (FTD).2 3 Most ALS cases are apparently sporadic (sALS), while in approximately 10% of patients, a positive family history can be identified (fALS).1 4 5 Although more than 50 potential ALS-related genes have been reported so far, a genetic aetiology may be determined in a minority of patients with sALS and in about two-thirds of fALS cases, with pathogenic variants in the SOD1, FUS and TARDBP, and repeat expansion in C9orf72 being the most common.4 6
Recent exome sequencing studies revealed TANK-binding kinase 1 (TBK1, MIM no 604834) as a novel causative gene of both ALS7 8 and FTD,9 suggesting haploinsufficiency as a disease mechanism. TBK1 encodes a multifunctional kinase protein that phosphorylates a wide range of substrates and exerts control over several cellular key processes, including innate immune response, inflammation, autophagy and cell proliferation.10 11 Notably, TBK1 interacts with optineurin (OPTN),12 valosin-containing protein and sequestosome-1 (SQSTM/P62),13 which have previously been reported as causative ALS genes.11
In this study, we have investigated the genetic role of TBK1 variants in a cohort of Italian patients with ALS and addressed their pathogenic potential using functional in vitro studies.
Materials and methods
Patients
The TBK1 gene was sequenced in a cohort of154 unrelated Italian patients with ALS. Blood samples were obtained for diagnostic purposes, after informed consent, and stored in our tissue bank; the study protocol was approved by the local ethic committee. ALS diagnosis was made according to El Escorial revised criteria.14 Patients with definite, probable, probable laboratory supported or possible ALS were included in the study. Although no strict inclusion or exclusion criteria were adopted, patients with earlier disease onset, fALS or cognitive impairment were prioritised for TBK1 sequencing by referring clinicians. Thirty patients (19.5%) had fALS,15 whereas 16 patients (10.4%) had a positive family history for FTD and 31 (20.1%) had cognitive impairment or unclassified dementia. The presence of pathogenic variants in known ALS genes was excluded by Sanger sequencing (SOD1, TARDBP and FUS/TLS) or repeat-primed PCR (C9orf72). The mean onset age of our cohort was 54.9±15.3 years (range 20–82 years), that is slightly younger compared with previous population-based studies.16 Eighty-seven patients (56.5%) were males and 67 (43.5%) were females. Hundred and sixteen (75.3%) had spinal-onset ALS and 38 (24.7%) had bulbar-onset ALS. The mean ALS Functional Rating Scale-revised (ALSFRS-R) Score at the first visit was 31.4±10.4 (range 4–47). All patients were of Caucasian ethnicity, except one who was of Hispanic origin.
Finally, we performed a PubMed literature review of all previously reported patients harbouring TBK1 variants and extracted, when available, their clinical and demographic characteristics.
Genetic analysis
For all patients, TBK1 whole coding region and exon junctions were analysed with a Sanger protocol, using the Big Dye Terminator V.1.1 Cycle Sequencing Kit (Applied Biosystems) (online supplementary table 1 for primers). Called sequences were aligned to the TBK1 reference sequence (National Center for Biotechnology Information (NCBI) Entrez gene ID 29110; NM_013254.3) with the Sequencer V.5.0 software (gene codes). Gene variants were evaluated by their absence or frequency in the public NCBI genome database, single-nucleotide polymorphism database (dbSNP), ExAc (Exome Aggregation Consortium) and EVS (Exome Variant Server). The Human Gene Mutation Database (HGMD) Professional 2016.4, the ALSoD (Amyotrophic Lateral Sclerosis Online Database) and PubMed have been interrogated to check for previously reported variants. The SIFT, PolyPhen-2 and MutationTaster in silico softwares were used to assess the functional effect of missense variants. Human Splicing Finder, NetGene2, neural network site, MaxEntScan and Gene Splicer were used to evaluate the potential effects on gene splicing. Crystal structure-based analysis was performed in order to asses the impact of novel TBK1 missense variants on protein thermodynamic stability (ΔΔG) and electrostatic surface potentials.17 18
Supplementary file 1
Transcript analysis and functional in vitro studies
Total RNA was extracted from patients’ peripheral blood samples, and transcript analysis was performed by RT-PCR to detect potential splicing defects of four TBK1 variants (online supplementary table 2 for primers). TBK1 mRNA and protein expression levels were quantified in patient-derived fibroblasts harbouring the c.358+5G>A variant, which were available for testing, by quantitative real-time PCR (qRT-PCR) and western blot. Expression, targeting and activity of the novel frameshift and missense TBK1 variants were studied in AD293 human embryonic kidney (HEK) cells and mouse motor neuron-like NSC-34 cells transfected with TBK1 plasmids containing the relevant mutations. Kinase activity of these variants was assessed by measuring the levels of phosphorylated interferon regulatory factor 3 (IRF3), a known substrate, while association with the canonical interactor OPTN was investigated with pull-down assays in transfected AD293 cells.
Extensive technical description is reported in online supplementary information.
Results
Identification of the novel TBK1 variants
Overall, we identified seven TBK1 variants in our cohort of unrelated Italian index patients (table 1). The p.Leu59Phefs*16 and the c.358+5G>A are absent from all public genomic databases, including dbSNP. The missense p.Lys291Glu and p.Arg357Gln, located in the kinase domain (KD) and ubiquitin-like domain (ULD), respectively, have been previously reported as displaying an impairment in TBK1 catalytic activity8 19 20 and thus regarded as functional missense mutations,20 potentially pathogenic. The missense p.Asp118Asn and p.Ile397Thr, as well as the intronic variant c.1644–5_1644-2delAATA are reported in the general population as variants at very low frequency (table 1). None of them has been previously described in association to a clinical phenotype. The Exac and EVS frequencies for the P.Lys291Glu, c.1644_5_1644-2delAATA and p.Ile397Thr are greater than expected for the disorder (table 1), thus lowering their probability to be pathogenic.
TBK1 variants identified in Italian ALS cases
The p.Leu59Phefs*16 frameshift results in a putative truncated protein of 73 amino acids. Likewise, as predicted by splice-site in silico softwares, the intronic variant c.358+5G>A is expected to completely abolish the recognition of the donor splice site, five base pairs downstream exon 4, leading to a putative aberrant protein product (online supplementary table 3). Thus, both mutations are predicted to be deleterious.
Conversely, in silico prediction analysis indicates that the intronic variant c.1644–5_1644-2delAATA does not affect the natural exon 15 acceptor site (online supplementary table 3). Moreover, this variant was also identified in the unaffected patient’s mother whose DNA was available for testing, not supporting its pathogenic role.
The missense variant p.Asp118Asn, located in the KD, is predicted as deleterious by PolyPhen-2 and Mutation Taster. Moreover, structure-based analysis confirmed that the presence of 118Asn determines significant conformational and electrostatic surface potential changes in the TBK1 catalytic domain, reducing protein global stability (ΔΔG: 3.98±0.27 kcal/mol) (online supplementary figure 1A,B). In contrast, the p.Ile397Thr variant, located in the linker region between the ULD and the coiled-coil domain 1 (CCD1), is predicted to be tolerated and benign by SIFT and PolyPhen-2, but predicted to be disease causing by Mutation Taster (table 1). Protein structure analysis showed that the mutant 397Thr has little effects on TBK1 structure and global stability (ΔΔG: 1.51±0.18 kcal/mol), with no changes in protein electrostatic surface potential (see online supplementary figure 1C,D). The p.Ile397Thr was absent in the patient’s unaffected sister, whose DNA was available for testing. Finally, since at the genomic level the coding sequences for p.Asp118Asn and p.Ile397Thr are located in the proximity of donor and acceptor splice sites, respectively (online supplementary table 3), an in silico splicing prediction was performed. This evaluation predicted a potential weak impact of the two variants, not supporting a relevant alteration of the splicing process.
Supplementary file 2
All the identified TBK1 variants were found in apparently patients with sALS, whose demographic and clinical characteristics are shown in table 2, while a summary of the main features of patients harbouring TBK1 gene variants reported so far is provided in table 3 (online supplementary table 4 for details).
Demographic and clinical characteristics of patients with TBK1 variants
Summary data of review of the literature of patients with TBK1 variants
Effect of variants on TBK1 transcript: the mutant c.358+5A allele disrupts processing of TBK1 transcript
In order to assess the potential consequences on splicing predicted in silico (online supplementary table 3), we performed a transcript analysis on patient’s peripheral blood samples. However, no aberrant transcripts were identified by RT-PCR and cDNA sequencing.
We were able to further investigated the intronic c.358+5G>A variant, since in silico analysis predicted inactivation of the canonical donor splice site (online supplementary table 3) could lead to exon 4 skipping, insertion of a premature termination codon (PTC) and degradation of the aberrant transcript through non-sense mediated decay (NMD) (figure 1A). In agreement with this hypothesis, quantitative RT-PCR analysis of TBK1 transcripts from blood and patient-derived primary fibroblasts showed a 50% reduction in total mRNA levels (figure 1B). Immunoblot analysis of patient’s fibroblasts confirmed a corresponding decrease in protein levels (figure 1D,E). Inhibition of NMD with cycloheximide restored normal levels of total TBK1 transcripts (figure 1B) and allowed detection of the aberrant isoform lacking exon 4 deriving from the mutated allele (figure 1C). Altogether, these results demonstrate that the c.358+5G>A genomic variant causes pre-mRNA misprocessing leading to TBK1 haploinsufficiency.
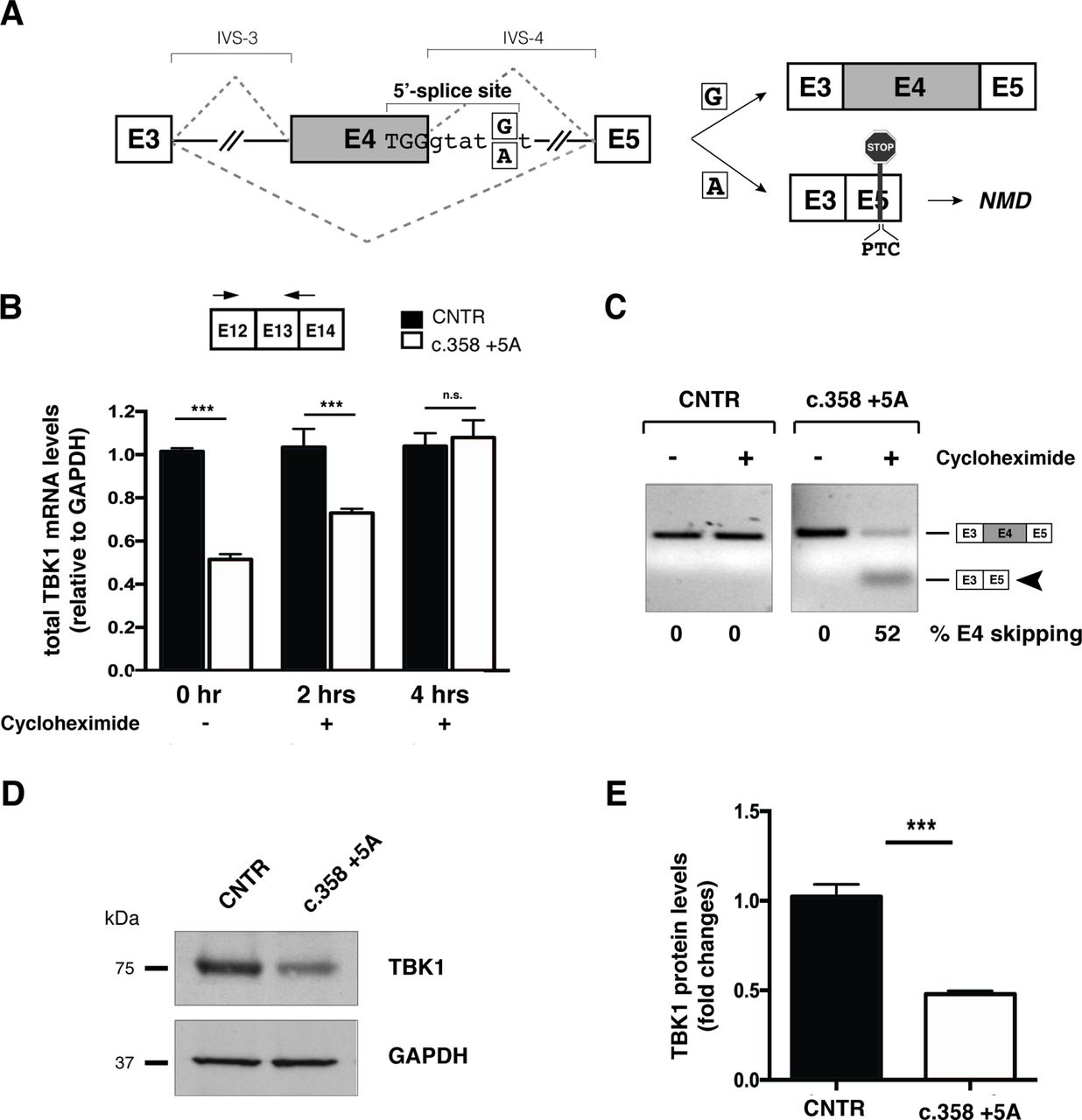
c.358+5A variant causes aberrant splicing of exon 4 leading to TBK1 haploinsufficiency. (A) Schematic representation of wild-type (c.358+5G) and mutated (c.358+5A) sequences. Exons are depicted as boxes (E3, E4 and E5) and introns (interVening sequence (IVS-3 and IVS-4) as lines. The sequence surrounding exon 4 donor splice site of the wild-type G and mutant A is indicated (position +5). The two possible splicing patterns (E4 inclusion or skipping) are indicated with diagonal dashed lines and the two possible resulting splicing products schematically reported on the right of the panel. (B) Analysis of total TBK1 mRNA levels by quantitative RT-PCR. Total RNA was extracted from primary control and c.358+5G/A fibroblasts at different time points (0, 2 and 4 hours) after cycloheximide administration. Arrows correspond to TBK1_E12/13 Fw and TBK1_E13 Rev quantitative PCR; primers. Results are expressed as mean±SD of three different experiments performed in triplicate. (C) TBK1 exon 4 splicing assay. Total RNA from samples in (B) was analysed by semiquantitative RT-PCR using TBK1_E2/3 Fw and TBK1_E5 Rev to detect inclusion or skipping (arrowhead) of exon 4 (see schematic diagrams in A). Band intensities were quantified and the percentage of exon 4 skipping is reported under each lane of the representative gel. (D) Immunoblot analysis of TBK1 protein levels in control and c.358+5G/A patient primary fibroblasts. GAPDH serves as a loading control. (E) Quantification of band intensity in (D) shows fold change reduction in c.358+5G/A fibroblasts relative to controls (mean± SD from three independent experiments). ***p-value <0.001. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; NMD, non-sense mediated decay; n.s., not significant (Student’s t-test); RT-PCR, real-time PCR; TBK1, TANK-binding kinase 1, CNTR: Control.
Functional characterisation of TBK1 variants
In order to assess the functional consequence of TBK1 variants, we introduced the p.Leu59Phefs*16 frameshift, the p.Asp118Asn and p.Ile397Thr missense variants in motor neuron-like NSC34 and HEK293T cell lines and studied their expression, targeting and activity. The p.Arg357Gln TBK1 missense, previously demonstrated as a potentially pathogenic functional missense mutation, was used as an experimental control.8
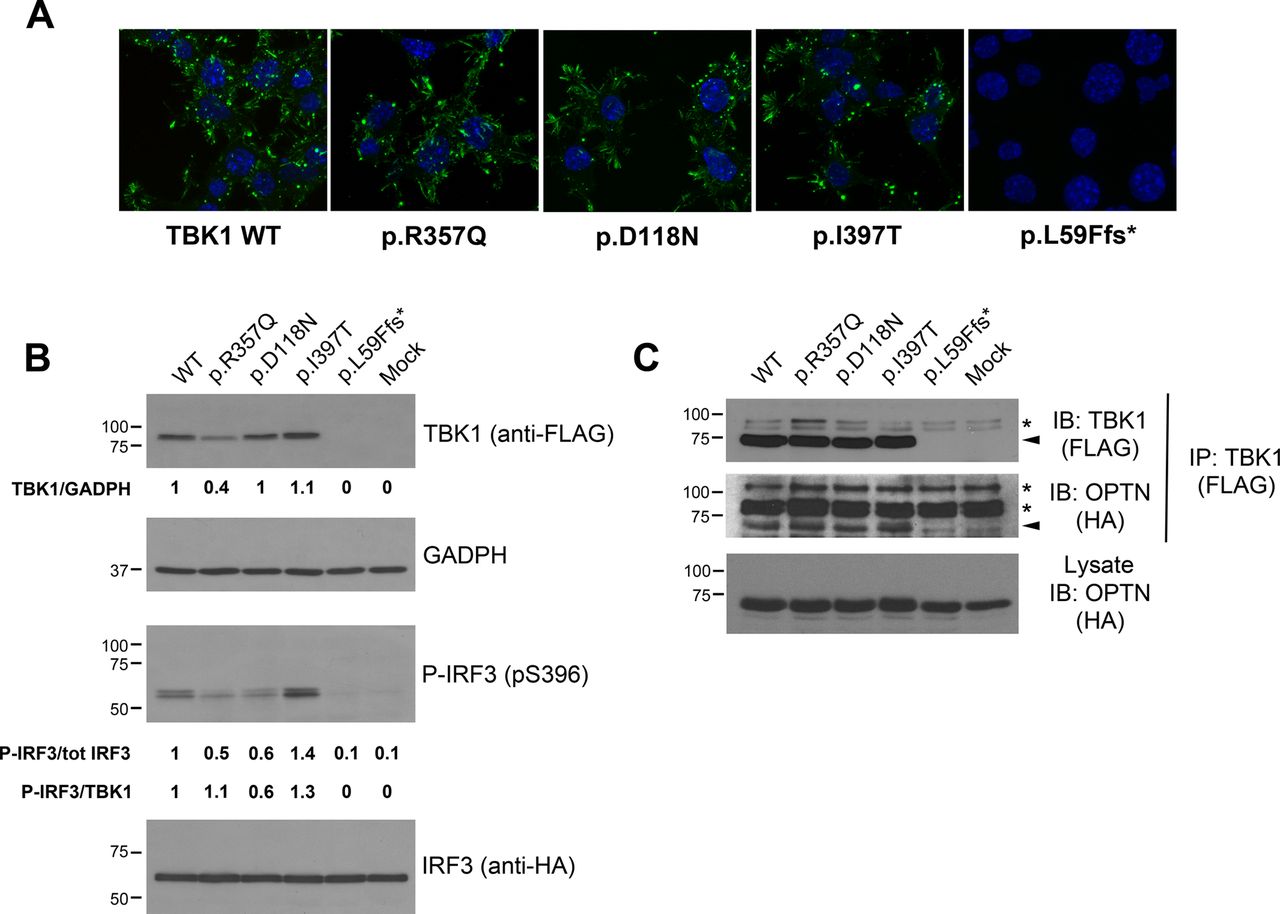
The p.Leu59Phefs*16 mutation leads to a putative truncated protein of 73 amino acids (8.5 kDa expected size) that was undetectable by immunofluorescence and western blotting (null allele) (figure 2A, B), whereas the p.Asp118Asn, p.Ile397Thr and p.Arg357Gln variants displayed a subcellular localisation virtually identical to wild-type TBK1 when expressed in motor neuron-like NSC34 cells (figure 2A). However, steady-state protein levels assessed by western blot were invariably lower for the p.Arg357Gln variant (~60% reduction) and approximately 10% higher for the p.Ile397Thr variant, relative to wild-type TBK1, suggesting an effect on protein turnover. On the contrary, the levels of the p.Asp118Asn TBK1 were comparable to those of the wild-type protein (figure 2B, top panels).

![[Supplementary_Figure_1.jpg]](https://jnnp.bmj.com/content/jnnp/88/10/869/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
{kind=link}
{kind=link}
In vitro functional characterisation of TBK1 missense variants. (A) Subcellular distribution of TBK1 mutants visualised by anti-FLAG staining in differentiated motor neuron-like NSC-34 cells expressing the indicated tagged constructs. Similarly to wild-type TBK1, mutants localise in the cytosol, vesicular structures and lamellar domains at the cell periphery. L59Ffs*16-TBK1 is undetected. (B) Expression levels and kinase activity of TBK1 mutants (Top panels). Western blot analysis of FLAG-tagged wild-type TBK1 and indicated variants expressed in human embryonic kidney (AD293) cells along with HA-tagged IRF3. Green fluorescent protein (GFP) transfected cells serve as control (mock). Quantification of steady-state protein levels of TBK1 mutants relative to wild-type and normalised to GADPH is reported below each lane. The steady-state protein levels are lower for the p.Arg357Gln and slightly higher for the p.Ile397Thr mutants. The truncating mutant p.Leu59Phefs*16 abolishes TBK1 expression. Phosphorylation of IRF3 on Ser-396 in the same protein lysates was revealed with a phospho-specific antibody (bottom panels). The ratio between p-IRF3 and total IRF3 is reported under the lanes (p-IRF3/tot IRF3) and these values are further normalised to the expression levels of each TBK1 mutant protein (p-IRF3/TBK1). Phosphorylation of IRF3 is impaired by the p.Asp118Asn mutation and enhanced by the p.Ile397Thr. (C) TBK1 mutants interact with OPTN. FLAG-tagged constructs of wild-type TBK1 and indicated missense variants (or GFP control plasmid; ‘mock’) were expressed in AD293 cells together with HA-tagged OPTN. TBK1 constructs were pull-down with anti-FLAG antibody; OPTN in the immunocomplexes was revealed with anti-HA antibody. Membranes were stripped and reprobed with anti-FLAG antibody to detect the immunoprecipitated TBK1 constructs. Arrows mark target proteins, whereas asterisks point to non-specific bands. (In the figure aminoacids are indicated with the one-letter code instead of the three-letter code). GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HA, haemagglutinin; OPTN, optineurin; p-IRF3, phosphorylated IRF3; TBK1, TANK-binding kinase 1; WT, wild type; IB: Immunoblot.
To determine whether these missense variants might interfere with the kinase activity of TBK1, we quantified the phosphorylation of its target IRF3 at Ser-396. TBK1 harbouring the p.Asp118Asn variant in the KD yields reduced levels of phospho-IRF3 (p-IRF3) relative to wild-type TBK1. Therefore, the p.Asp118Asn can be regarded as a functional missense mutation, potentially pathogenic. In contrast, p-IRF3 levels were elevated in cells expressing the p.Ile397Thr variant, which is located in the linker region between the ULD and CCD1 domains (figure 2B, bottom panels). Interestingly, p-IRF3 levels were approximately 50% lower in cells expressing the p.Arg357Gln variant, located in the ULD, consistent with previous reports8; however, this decrease was related to a substantial reduction in the steady-state levels of this variant (figure 2B, top panels).
We finally examined the ability of TBK1 variants to interact with the autophagy receptor OPTN. Pull-down assays with tagged constructs revealed that the three TBK1 missense mutants (p.Asp118Asn, p.Ile397Thr and p.Arg357Gln) were competent for association with OPTN, whereas the p.Leu59Phefs*16, as expected, did not bind OPTN (figure 2C).
Discussion
In order to understand the relevance of TBK1 as a disease gene in the Italian ALS population, we analysed a cohort of 154 unrelated patients with ALS by sequencing the whole coding region and exon junctions. We identified two loss of function (LoF) variants, the frameshift p.Leu59Phefs*16 and the intronic c.358+5G>A, never reported before. In addition, we found two missense, p.Arg357Gln and p.Lys291Glu, previously described in one patient with ALS and one patient with FTD, respectively,8 19 as well as two missense, p.Asp118Asn and p.Ile397Thr, and one intronic variant, c.1644–5_1644-2delAATA, never associated with a clinical phenotype or ALS and reported at low frequency in the general population.
A number of TBK1 LoF pathogenic variants8 has been described so far, severely reducing the protein levels and suggesting a pathogenic mechanism based on haploinsufficiency.9 19 21 In the present study, using functional in vitro assays we showed that the p.Leu59Phefs*16 frameshift results in undetectable TBK1 protein product, with consequent loss of both its catalytic function and binding ability. Also for the novel splice site c.358+5G>A, we were able to demonstrate a loss of function since this variant resulted in exon 4 skipping, introduction of a PTC and ultimately to a 50% loss of transcript and protein levels in the patient, heterozygous for this variant. Interestingly, the previously described LoF variant, c.358+2T>C, located in the same donor splice site, was similarly not expressed at the protein level, but able to induce NMD through a different mechanism, namely intron 4 retention.8 The p.L59Ffs*16 and c.358+5G>A variants can be, therefore, considered as novel LoF pathogenic mutations, leading to TBK1 haploinsufficiency.
In addition to LoF mutations (null alleles), TBK1 functional impairment may also result from missense variants. Indeed, mutations in the KD may affect the protein catalytic activity, whereas mutations in the CCD1 may impair association with binding partners, like OPTN, even though it is still not clear which functions of TBK1 are relevant for neurodegeneration.21 Here, we showed that the novel p.Asp118Asn variant, located in the KD, exhibited reduced levels of phosphorylation activity without affecting unchanged protein levels, consistently with our structure-based analysis results and supporting its role as a potentially pathogenic functional missense mutation. This conclusion was also supported by the observed high evolutionary conservation of the Asp118 codon, up to zebrafish. Similarly, we confirmed that previous findings for the p.Arg357Gln missense variant, located in the ULD, were able to impair TBK1 catalytic activity.8 Intriguingly, this reduction seemed related to a decrease of TBK1 protein levels rather than an intrinsic catalytic defect. Indeed, impairment of the catalytic activity might also be due to a combination of both loss-of-protein and protein function, which might in turn be due to instability of the mutated protein, a mechanism previously hypothesised for the p.L94S missense mutation.20 In contrast, the p.Ile397Thr variant exhibited a substantial increase in TBK1 phosphorylation activity, partly associated with higher protein levels. We speculate that the p.Ile397Thr variant may trigger an alternative dominant pathogenic mechanism involving enhanced TBK1 kinase activity and/or reduced protein turnover that might be as disruptive for cellular metabolism as the decrease caused by other pathogenic TBK1 mutations. However, the potential biological relevance of an increase of TBK1 phosphorylation activity or an imbalance of TBK1 protein turnover is not clear with respect to ALS/FTD pathogenesis and requires further investigation.
Our data support the wide heterogeneity of TBK1 related phenotypes, consistently with previous findings.20 22–24 Age of onset in our mutated patients ranged from 36 to 81 years. Therefore, TBK1 mutations may also be considered in early-onset patients with ALS. Moreover, while all our patients had spinal onset, a wide spectrum of clinical phenotype could be observed, ranging from classic ALS to flail arm and pure lower motor neuron ALS. Cognitive impairment was observed in three patients,25 supporting the role of TBK1 mutations in the ALS–FTD spectrum and further emphasising the concept that ALS and FTD share a common molecular and genetic background.8 19
Overall, in our cohort of Italian patients with ALS the observed frequency of TBK1 LoF mutations was 1.3% (2/154), increasing up to 3.2% (5/154) by taking into account also the functional missense variants that we were able to classify as potentially pathogenic. This frequency is comparable with respect to previous ALS cohorts8 19 20 26 and slightly higher in comparison to that reported in a Sardinian isolated population (1.6%).22 However, we acknowledge as a limitation that this is not a population-based study; therefore, the genetic epidemiology of TBK1 variants in the Italian ALS population requires further confirmation. Interestingly, in our cohort, LoH mutations were detectedin two 36-year-old woman, suggesting a potential gender effect. However, this hypothesis is not supported by a review of the literature of all previously reported cases, showing a balanced male to female ratio and no relevant gender effect on age of onset. TBK1 pathogenic variants were found in apparently patients with sALS, whereas no variants were detected in our fALS population; however, two patients had a positive family history for unclassified dementia suggesting a potential intrafamiliar overlap within the FTLD (frontotemporal lobar degeneration) spectruNotably, our patients harbouring the already reported p.Arg357Gln8 and p.Lys291Glu19 20 functional missense variants presented with a phenotype differing from previously described patients with regard to age, site of onset and disease duration. Furthermore, the p.Lys291Glu, located in the KD and able to impair TBK1 phosphorylating activity, has been previously reported in one patient with FTD and also in two control subjects.19 20 Altogether, these findings support the hypothesis that TBK1 pathogenic variants have a reduced penetrance and may be associated with a considerable phenotypic heterogeneity within the ALS/FTD spectrum, as also observed in multigenerational pedigrees segregating a TBK1 mutation,8 19 20 27 and similarly to what is described for C9orf72 repeat expansions.28 However, a contribution of de novo mutations, compound heterozygous variants or genetic variants in multiple genes, consistent with an oligogenic model of disease may be taken into consideration.1 29
In conclusion, although the specific pathomechanism driven by TBK1 is still poorly understood, functional studies can provide critical evidence for the pathogenicity of unreported TBK1 variants, especially when segregation studies are not feasible. This study supports the relevance of TBK1 gene mutations in the Italian ALS and ALS–FTD spectrum population, suggesting its inclusion in the panel of genes to be sequenced in ALS.
Acknowledgments
The authors thank Linda Ottoboni and Francesca Ruffini for their technical assistance.
Acknowledgments
The authors thank Linda Ottoboni and Francesca Ruffini for technical assistance.
References
Footnotes
Contributors LP, FV, LM, ADM, TD, AR and CT performed the experiments and acquired the data; LP, FV, LM, ADM, TD, AR, FA, SP, DB, PC and NR contributed to analysis and interpretation of the data; YMF, LT, GS, FC, RF, FA, CL and NR recruited the patients, performed the clinical evaluation, acquired clinical data and collected patient DNA; LP, FV, ADM, AR, YMF, DB, PC and NR drafted and revised the manuscript; LT, GS, FA, GC, MF, AQ, CL, SP, DB, PC and NR critically revised the manuscript for intellectual content; AQ, CL, SP, DB, PC and NR conceptualised and designed the study, and interpreted the analysis; FA, GC, MF, AQ, CL, SP, DB, PC and NR obtained the funding; AQ, SP, DB, PC and NR supervised the study; all authors had full access to the data in the study and approved the final version of the manuscript.
Funding This work was supported by AriSLA, Italian Research Foundation for ALS (ExAlta Grant) to AQ, the Giovanni Armenise-Harvard Foundation and ERC-StG 35590–NEVAI to DB and the Italian Ministry of Health (grant RF-2010-2313220) to AF.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The study was approved by the local Ethical Committee of the San Raffaele Scientific Institute.
Provenance and peer review Not commissioned; externally peer reviewed.