Article Text

Abstract
Neuroendocrine abnormalities are common in Parkinson's disease (PD) and include disruption of melatonin secretion, disturbances of glucose, insulin resistance and bone metabolism, and body weight changes. They have been associated with multiple non-motor symptoms in PD and have important clinical consequences, including therapeutics. Some of the underlying mechanisms have been implicated in the pathogenesis of PD and represent promising targets for the development of disease biomarkers and neuroprotective therapies. In this systems-based review, we describe clinically relevant neuroendocrine abnormalities in Parkinson's disease to highlight their role in overall phenotype. We discuss pathophysiological mechanisms, clinical implications, and pharmacological and non-pharmacological interventions based on the current evidence. We also review recent advances in the field, focusing on the potential targets for development of neuroprotective drugs in Parkinson's disease and suggest future areas for research.
- PARKINSON'S DISEASE
- DIABETES MELLITUS
- METABOLIC DISEASE
- NEUROENDOCRINOLOGY
- SLEEP DISORDERS
Statistics from Altmetric.com
Introduction
Parkinson's disease (PD) is a common neurodegenerative condition characterised by motor and non-motor symptoms (NMS). While the classic motor features are attributable to nigrostriatal dopaminergic cell loss, the spectrum of NMS reflects a more complex aetiology including neuroendocrine and metabolic abnormalities.
Neuroendocrine abnormalities in PD are important for several reasons. They are common, mainly recognised and studied in advanced stages of PD, and associated with multiple NMS.1 However, they appear to be an integral feature of PD at all stages of disease, not secondary to disruption of other physiological processes or side effects from medication. Recent advances have shed light on the underlying pathophysiology and relationship to PD, although important questions remain regarding the effect of neurodegeneration on neuroendocrine axes. A better appreciation of the neuroendocrine abnormalities in PD and their clinical implications may allow tailored clinical assessments and offer better symptomatic therapeutic interventions. In addition, neuropeptides and hormones are easy to assay in various body fluids (blood/urine/saliva). Altered concentrations may correlate with disease severity and play a role in disease progression and pathogenesis. As such, they represent potential biomarkers of disease state. Finally, neuroendocrine abnormalities could form the basis for the future development of targeted therapies for NMS and neuroprotective treatments in PD.
This review does not cover every endocrine system or metabolic abnormality reported to be disrupted in PD, but provides a systems-based overview of those where there have been recent advances in terms of the clinical or therapeutic implications. We discuss the current epidemiological and pathophysiological evidence available, future areas for research, and give therapeutic recommendations for each of these neuroendocrine and metabolic disorders in PD.
Search strategy
A PubMed/MEDLINE search was performed for articles published in English between January 1990 and June 2016. We combined searches using ‘Parkinson's disease’ and the keywords ‘neuroendocrine’, ‘circadian disorder’, ‘suprachiasmatic nucleus’, ‘hypothalamus’, ‘melatonin’, ‘pineal gland’, ‘diabetes’, ‘insulin resistance’, ‘glucose intolerance’, ‘body weight’, ‘feeding behaviour’, ‘leptin’, ‘ghrelin’, ‘osteoporosis’, ‘bone mineral density’ and ‘vitamin D’. Reference lists were manually checked to capture any additional articles. The final list of references was generated based on the relevance of the articles to the aim of this review.
Circadian rhythm and sleep disorders
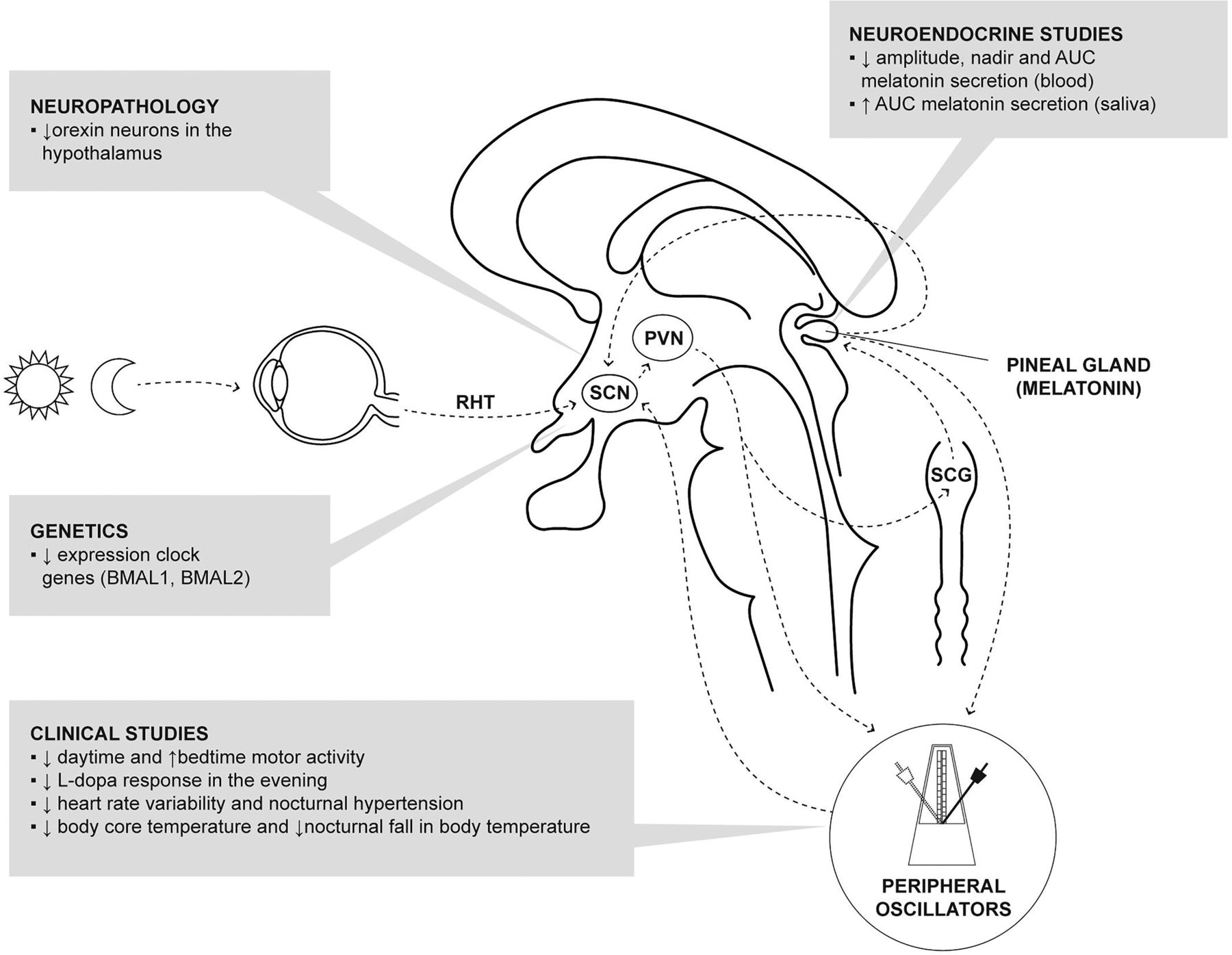
Circadian (daily) rhythms are present in almost all physiological systems of the human body, the sleep–wake cycle being most apparent. The system responsible for this near 24-hour rhythm is composed of a central pacemaker and peripheral oscillators (figure 1). The central biological master clock is located in the suprachiasmatic nucleus (SCN) of the anterior hypothalamus2 and its rhythmic activity is the result of the expression of clock genes. The SCN is entrained to the 24-hour environmental light cycle through the retinohypothalamic tract, circulating melatonin and time cues from peripheral oscillators. Melatonin is the most important endogenous entraining agent and its production by the pineal gland during darkness is regulated by the SCN.3

Circadian system and its dysregulation in Parkinson's disease. The SCN of the hypothalamus is the central pacemaker and its rhythmic activity is the result of the expression of clock genes. The SCN receives photic information from the retinohypothalamic tract, cues from peripheral oscillators and circulating melatonin. It also regulates melatonin secretion via an indirect multisynaptic pathway reaching the pineal gland via the PVN of the hypothalamus and the SCG. Main disruptions found in PD are shown in shaded boxes. AUC, area under the curve; PD, Parkinson's disease; PVN, paraventricular nucleus; RHT, retinohypothalamic tract; SCG, superior cervical ganglion; SCN, suprachiasmatic nucleus.
Coordination of circadian rhythms is an essential element of optimal physical and mental health4 and its disruption has been associated with metabolic disturbances,5 disorders of the immune system,6 increased cancer risk,7 renal dysfunction,8 cardiovascular disease,9 impaired cognition,9 psychiatric and mood disorders.10 ,11 Growing evidence suggests that alterations of the circadian system in patients with PD might contribute not only to sleep–wake cycle dysregulation but also to other NMS.
Pathophysiology and abnormalities in PD
Clinical circadian abnormalities
Motor function: actigraphic studies have demonstrated disruption of the physiological motor pattern, with patients with PD displaying increased activity at bedtime and reduced activity levels during the day which correlates with disease stage.12 ,13 Moreover, patients with PD exhibit worsening of their motor symptoms with diminished motor response to levodopa therapy in the evening unexplained by pharmacokinetic factors14 ,15 which may reflect disruption of circadian regulation of dopaminergic systems.16
Non-motor function: cardiovascular circadian rhythms are also disrupted in PD, with reduced heart rate variability12 ,17 ,18 and reversal of the circadian blood pressure profile with nocturnal hypertension.19 ,20 A lower core body temperature and reduced nocturnal fall in body temperature have also been reported, suggesting a circadian disruption of thermoregulation.21 Patients with PD also show circadian fluctuations of visual performance measured in contrast sensitivity,22 linked to altered diurnal fluctuations in retinal dopamine.23 Although other elements including autonomic dysfunction and the effect of medication are likely to have an impact, these studies have demonstrated disrupted circadian rhythms contributing to these abnormalities.
Sleep: sleep disorders in PD are very common and include sleep fragmentation, insomnia, REM sleep behaviour disorder (RBD), restless legs syndrome and excessive daytime sleepiness.24 They have a multifactorial origin including re-emergence of motor and NMS at night, nocturia, side effects of dopaminergic and other medications, and alterations in the circadian regulation of the sleep–wake cycle. Sleep disturbances in PD have been correlated with increased α-synuclein load and neurodegeneration of brain regions involved in promoting sleep such as the lower brainstem (locus coeruleus, raphe nuclei), amygdala, thalamus and hypothalamic (paramammillary and posterior nuclei).25 The wake-promoting effect of the orexin system of the lateral hypothalamus has also been implicated in the pathogenesis of sleep disturbances in PD. Cerebrospinal fluid (CSF) orexin levels in PD have shown conflicting results depending on CSF sampling site (ventricular vs lumbar) and stage of the disease,26–28 but well-designed pathological studies showed a severe reduction of orexin neurons in the lateral hypothalamus correlating with disease severity.29 ,30 In addition to these neuroanatomical structures, disruption to the molecular elements of the circadian system (see below) are believed to contribute to sleep disorders in PD.
PD pathophysiology: study of animal models has suggested that alterations in the circadian system might accelerate the pathological processes underlying PD.31
Functional circadian abnormalities
Clock genes: at a molecular level, circadian rhythms are regulated by several clock genes forming a set of interlocking transcription–translation feedback loops. Their pattern of expression has been proposed as a peripheral marker of circadian activity.32 Abnormalities of clock genes in peripheral blood of patients with PD include altered expression of Bmal1,33 ,34 Bmal235 and altered promoter methylation of Npas2.36 However, the clinical implications associated with these changes are unclear.
Melatonin: as there is no pineal storage of melatonin, circulating concentrations are considered a good biological marker of the circadian system.3 Early studies showed a phase advance of the nocturnal melatonin secretion, and decrease in night-to-daytime ratio of melatonin secretion which probably reflects dopaminergic treatment.37 ,38 Recent studies, with careful design to control the effects of exogenous variables, showed diminished amplitude of serum melatonin secretion in patients with PD on dopaminergic therapies, which correlated with excessive daytime sleepiness39 and various alterations in sleep architecture.34 In contrast, an increase in salivary melatonin was found in treated, but not in patients with unmedicated PD or controls.40 Differences in experimental protocols (particularly sample type, sample collection timing and control of exogenous factors) makes comparison between these studies challenging.
Cortisol: the secretory rhythm of cortisol is a sensitive marker of circadian function and persistently elevated concentrations of cortisol in blood34 ,41 and saliva42 have been reported in patients with PD. However, while the recognised effect of exogenous stress on cortisol concentrations makes interpretation difficult, impulse control behaviours,42 weight changes after deep brain stimulation (DBS)43 and mood disturbances44 have all been associated with cortisol secretion abnormalities in patients with PD.
These preliminary data suggest that circadian rhythm disruption is an early feature of PD as these abnormalities were found in newly diagnosed patients34 although the neuroanatomical site of disruption remains unclear. A recent study showed a reduction in hypothalamic grey matter volume (measured using MRI) in patients with PD compared with controls, together with a linear correlation between hypothalamic volume and 24-hour melatonin output in the PD group.45 Since melatonin is produced by the pineal gland under circadian control, collectively these results suggest that degenerative changes in neural structures controlling pineal output (such as the SCN) may be responsible for reduced melatonin output in PD. Further study of neuroanatomical components regulating the circadian system (eg, the pineal gland) should be performed in PD.
Therapeutic implications
Melatonin: Dowling et al46 compared the administration of melatonin 5 or 50 mg/day versus placebo for a period of 2 weeks in a randomised controlled cross-over trial of 40 patients with PD with sleep disturbances. Actigraphy showed a minimal increase in total night-time sleep (10 min) in the high-dose group, but only subjective improvement in sleep quality in the lower dose group, compared with placebo. Another study with 18 patients with PD randomised to melatonin 3 mg/day or placebo for 4 weeks showed significant improvement in subjective quality of sleep in the melatonin group but no significant differences on polysomnography.47 Based on these results,48 a consensus from the Movement Disorder Society concluded that there was insufficient evidence to recommend the routine use of melatonin for the treatment of insomnia in PD.49 Further studies with large samples, longer duration, careful protocol design to control exogenous factors and identification of patient subgroups where sleep abnormalities are likely to be secondary to circadian dysfunction are warranted.
Bright light therapy (BLT): It has been postulated that BLT might restore circadian rhythmicity in PD, as it has demonstrated efficacy in the treatment of mood disorders.50 Although promising results have been reported on its effect on sleep, mood and motor function in patients with PD,51 BLT has only been assessed in a few studies with different light therapy regimes and assessment protocols, making it difficult to draw firm conclusions. The only randomised placebo-controlled trial of 18 patients with PD treated with BLT showed improvement of mood disturbances, parts I, II and IV of the Unified Parkinson's Disease Rating Scale (UPDRS) in comparison to the placebo group, but failed to improve motor UPDRS or sleep.52 Other cases series53 and retrospective open-label studies54 have shown additional improvement of sleep and motor function. Further studies with standardised protocols and rigorous design are required to validate these results.
Key points
Studies have shown evidence for disruption of circadian rhythms in motor and non-motor activities, and of markers of circadian activity (clock genes, melatonin, cortisol) in patients with PD.
This is likely to reflect disruption of the circadian system at many levels including the activity of the SCN and its humoral outcome signal melatonin.
Although melatonin and BLT could be potential treatment options for these circadian disruptions, further evidence is needed to justify their use.
Diabetes and glucose metabolism
The potential association between PD and type 2 diabetes mellitus (T2DM) has long been recognised,55 and has been the subject of increased research attention in recent years.56
Epidemiology
The prevalence of glucose intolerance has been estimated to be as high as 80% in patients with PD in historical studies,55 although recent epidemiological data are conflicting. A recent meta-analysis of case–control studies reported a negative association (OR=0.75 (95% CI 0.58 to 0.98))57 although still observed that 2.9% of patients with PD had a diagnosis of diabetes compared with only 1.6% of non-PD population. Case–control studies are potentially prone to selection bias towards individuals attending specialist clinics, and cannot account for subsequent development of either PD or diabetes, making findings of association tentative. Indeed these results contrast with a meta-analysis of prospective studies, in which pre-existing T2DM was found to be a risk factor for future PD (RR=1.26 (95% CI 1.03 to 1.55); p<0.0001).58 ,59 Conflicting results might be explained by heterogeneity between studies, differing case ascertainment of both conditions, the potential for misdiagnosis, and failure to take into account for the modulating effect of diabetes medications on disease expression. Environmental and ethnic factors might also affect the association in different populations.
T2DM might also exert a modifying effect on PD phenotype and disease progression. A case–control study showed that patients with PD with antecedent diabetes had more severe motor symptoms, higher scores on the motor UPDRS, and required higher doses of levodopa.60 Clinical studies have shown that the presence of T2DM is associated with specific phenotypes, including greater postural instability, gait difficulties and cognitive impairment.61–63 This association is clinically relevant since axial motor symptoms and cognitive impairment are less responsive to dopaminergic therapies and are a major cause of disability. The lack of therapeutic response might be secondary to non-dopaminergic neurotransmitter involvement, as the phenotypic variability was not explained by differences in nigrostriatal dopaminergic denervation on (11C)dihydrotetrabenazine positron emission tomography (PET) scans in patients with PD with and without T2DM.61
Pathophysiology and abnormalities in PD
Recent studies have provided potential mechanisms by which T2DM could be a risk or modifying factor for PD:
Cerebrovascular disease: increased prevalence of vascular pathology and vascular parkinsonism in patients with T2DM might account for these findings. However, epidemiological association between PD and T2DM remained significant after adjustment for vascular risk factors and exclusion of participants with clinical cerebrovascular disease.58 ,59 MRI studies of the presence of cerebrovascular disease and leukoaraiosis showed no differences between groups of patients with PD with or without diabetes.61
Dopaminergic medication: the effect of some anti-PD medications on glucose metabolism has been suggested as a potential confounding factor, since evidence suggests a reciprocal regulation between insulin and brain dopaminergic activity.64 Chronic treatment with levodopa has been shown to induce decreased glucose tolerance, hyperglycaemia and hyperinsulinaemia.65 ,66 On the other hand, bromocriptine increases insulin sensitivity, improves glycaemic control and is licensed for the treatment of diabetes.67 However, reduced insulin-mediated glucose uptake,65 and inhibition of early insulin secretion and long-term hyperinsulinaemia and hyperglycaemia after glucose loading68 have also been found in samples of drug-naïve patients, supporting the hypothesis that abnormal insulin signalling and glucose metabolism predate dopaminergic treatment in patients with PD.
Cellular and molecular biology: it has been hypothesised that aberrant insulin signalling might ultimately lead to insulin resistance and diabetes, and put individuals at increased risk for PD.56 ,69 Mitochondrial dysfunction, neuroinflammation, increased endoplasmic reticulum stress, abnormal protein aggregation and metabolic abnormalities are common to both diabetes and PD, suggesting a pathophysiological link.56 ,69
Therapeutic implications
The common pathophysiological mechanisms shared by T2DM and PD may lead to more effective treatments which target both conditions. A prospective observational study showed that treatment using a combination of metformin and a sulfonylurea appeared to have a protective effect on the risk of developing PD in a Taiwanese cohort of patients with diabetes.70 Special attention has focused on the potential neuroprotective properties of peroxisome proliferator-activated receptor γ (PPAR-γ) and its coactivator 1-α (PGC1α) due to its pivotal role in mitochondrial respiration and gluconeogenesis. The thiazolidinediones (such as pioglitazone and rosiglitazone) are a class of PPAR-γ agonist. They have been successfully tested for their neuroprotective potential in animal models of PD.71 The potential therapeutic effect of these drugs on PD was further supported by a retrospective cohort study which showed a 28% lower rate of developing PD in those patients with diabetes treated with thiazolidinediones compared with other antidiabetic drugs.72 These results prompted a large, multicentre, double-blind, placebo-controlled trial including 210 patients randomly assigned to 45 mg/day pioglitazone, 15 mg/day pioglitazone or placebo to assess the potential effect on patients with PD. Results failed to show a significant benefit on symptoms (measured using total UPDRS) and the authors concluded that pioglitazone was unlikely to modify clinical progression in PD at the doses studied.73
More promising are the preliminary clinical results for exenatide, a synthetic glucagon-like peptide 1 (GLP1) receptor agonist licensed for the treatment of diabetes, which has been evaluated as a neuroprotective agent in patients with PD (for a detailed description of PD pathogenesis and GLP1 receptor stimulation, see review by Athauda and Foltynie).74 An initial open-label randomised controlled trial comparing 20 patients with PD treated with exenatide and 24 patients with PD acting as controls showed a clinically relevant improvement in motor (5.6 points on part 3 UPDRS) and cognitive (5.3 points on Mattis Dementia Rating Scale) domains in the treatment group after 12 months.75 Further studies with larger samples are currently on going (ClinicalTrials.gov number NCT01971242).
Key points
Pre-existing T2DM appears to be a risk factor for PD and to modify its clinical course with more severe disease progression, axial motor symptoms and cognitive impairment.
The pathophysiological link is not well understood but it may involve mitochondrial dysfunction, neuroinflammation, increased oxidative stress, abnormal protein aggregation and metabolic abnormalities
Owing to shared pathophysiology, several antidiabetic drugs are being explored as potential treatment for PD with promising initial results in the case of exenatide.
Body weight and energy metabolism
Extensive research on the mechanisms governing body weight, feeding behaviour and energy metabolism has provided insight into complex interactions between peripheral signals and the central nervous system. The classic concept of anatomically distinct ‘satiety/feeding’ centres has been gradually replaced by a more complex network of interconnected neurons of homoeostatic and hedonic systems, receiving and integrating multiple orexigenic and anorexigenic signals from peripheral tissues, nutrients and other areas of the central nervous system (figure 2).76–78

Feeding behaviour regulatory mechanisms and their dysregulation in Parkinson's disease. Feeding behaviour is regulated by complex interactions between homoeostatic and hedonic mechanisms. The hypothalamus is the central component of the homoeostatic control and regulates the anorexigenic and orexigenic activity of its neurons using the information from peripheral signals. Main disruptions found in PD are shown in shaded boxes. Orexigenic areas are represented in hatched ovals and anorexigenic areas in white ovals. AgRP, agouti-related protein; BMI, body mass index; CART, cocaine and amphetamine-regulated transcript; CCK, cholecystokinin; DBS, deep brain stimulation; INF, infundibular nucleus; LHA, lateral hypothalamic area; MCH, melanin-concentrating hormone; MSH, melanocyte-stimulating hormone; NA, nucleus accumbens; NPY, neuropeptide Y; OXM, oxyntomodulin; PD, Parkinson's disease; VTA, ventral tegmental area.
The hypothalamus is the central component of the homoeostatic control of feeding behaviour with anorexigenic and orexigenic cells: the infundibular nucleus produces cocaine and amphetamine-regulated transcript and melanocyte-stimulating hormone with anorexigenic activity, while the orexigenic cells are located in the infundibulum via neuropeptide Y (NPY) and agouti-related protein neurons and lateral hypothalamic area (orexin and melanin-concentrating hormone—MCH). The activity of hypothalamic neurons is influenced by peripheral humoral signals with opposite functions including leptin, ghrelin, gut satiety peptides and also levels of insulin, glucose or fatty acids. Leptin is an adipokine synthesised by fat tissue reflecting the energy reserve and produces anorexigenic effects, whereas ghrelin, a peptide synthesised by the gastric mucosa during fasting, promotes feeding, weight gain and stimulates growth hormone secretion. The hedonic control (sensorial information, food reward systems) is integrated in several areas including the mesolimbic dopaminergic system, insular cortex, dorsal striatum, and anterior cingulate and orbitofrontal cortices.
Epidemiology
The mechanisms regulating food intake might be implicated in other behaviours and brain functions including learning and memory, and the positive association between obesity, brain atrophy and dementia is recognised.79 A causal relationship between being overweight and PD is more controversial and results from prospective epidemiological studies are inconclusive.80 Some have shown a positive association of indices of obesity (body mass index (BMI)81–83 and triceps skinfold thickness)84 with an increased risk of PD, although these results have not been reproduced in other cohorts.85 ,86
On the other hand, the inverse association is well recognised and unintentional weight loss has been consistently reported with PD (affecting ∼50% of patients).82 ,87–89 A meta-analysis including 871 patients showed an overall reduction of 1.73 kg/m2 in patients with PD compared with controls, with a positive association with disease severity but not with disease duration.90 This weight loss carries important clinical implications and appears to be associated with a more rapid disease progression91 and to correlate inversely with health-related quality of life.92
Pathophysiology and abnormalities in PD
PD intrinsic factors
Dopaminergic dysfunction: owing to the role of dopamine in the regulation of the hedonic mechanisms of feeding behaviour,93 dopamine dysfunction producing anorexigenic signals in the hypothalamus has been proposed to contribute to weight loss in PD.
Levodopa: despite the fact that weight loss has been shown to be more prominent after starting levodopa treatment in observational studies,94 ,95 it seems that the levodopa requirement simply reflects disease severity. In addition, weight loss in PD has been reported before treatment with dopaminergic therapies,96 sometimes predating the onset of motor symptoms.88
Energy expenditure/intake imbalance: reduced caloric intake secondary to motor (rigidity, impaired hand coordination) and gastrointestinal (dysphagia, reduced bowel motility, upper gastrointestinal symptoms) complications have been proposed as a factor driving the energy imbalance contributing to weight loss in PD. However, several studies have demonstrated that weight loss occurs despite an increased energy intake in patients with PD.88 ,96 Given the correlation between weight loss and disease severity,90 ,97 motor symptoms (tremor, rigidity) and motor complications (dyskinesias) could potentially increase the energy expenditure at rest resulting in weight loss.98 However, other studies have demonstrated that the total daily energy expenditure is not higher in patients with PD with weight loss compared with patients with PD without weight loss99 and healthy controls,100 arguing against the possibility that abnormally elevated energy expenditure contributes to weight loss in PD. Overall it seems that the weight loss in PD is not explained by an energy imbalance, and can occur despite an increase in caloric intake.
Peripheral mechanisms of feeding behaviour regulation
Leptin: measurement of leptin97 ,101 and other adipokines102 have shown no significant differences between patients with PD with and without weight loss, and controls. Despite results showing a trend towards reduced concentration in patients with PD, this correlates with BMI and is likely that reduced leptin concentration reflects reduced body fat tissue content rather than being a causal factor for weight loss.
Ghrelin: ghrelin levels rise with prolonged fasting and fall rapidly after food ingestion, with an overall negative correlation with body weight. In patients with PD, however, there is a lower plasma ghrelin concentration in those patients with lower BMI103 and a reduction in the levels of ghrelin after the postprandial fall in patients with PD and idiopathic RBD,104 suggesting dysregulation of its secretion. Since RBD is considered a potential premotor stage of PD, ghrelin has been proposed as a potential peripheral biomarker for early PD.104 Recent studies demonstrated that ghrelin exerts a number of roles in other extrahypothalamic tissues including activation of the dopaminergic nigrostriatal system, hippocampus and mesolimbic dopaminergic system, and is implicated in learning and memory, reward behaviour, motivation, anxiety and depression.105 More importantly, ghrelin is reported to have neuroprotective properties in the nigrostriatal system in experimental animal models of PD106 mediated by the same mitochondrial function regulator (PGC1α)107 suggested as a potential therapeutic target in neuroprotection for PD and T2DM.56 Although these findings need to be replicated in humans, ghrelin appears a possible therapeutic target for disease neuroprotection, as well as treatment target for NMS such as obesity, apathy and depression.
Central mechanisms of feeding behaviour regulation
DBS: the role of the central regulatory hypothalamic mechanisms in weight disturbance in PD has recently attracted much attention in part due to the effects of DBS on body weight. Rapid weight gain has been consistently reported in multiple studies of patients with PD after subthalamic nucleus (STN) DBS. This greatly exceeds the weight loss seen in medically treated patients.108–111 These effects are not observed in patients with essential tremor undergoing DBS of the motor thalamus.112 Various mechanisms have been postulated, but it seems that STN DBS may induce changes in the regulatory mechanism of the hypothalamus with normalisation of energy metabolism.113 These effects seem target-dependant, being more marked with bilateral STN stimulation (compared with unilateral STN stimulation114 or globus pallidus internus stimulation)115 and with more medially placed electrodes in STN DBS.116 A stimulatory effect of the DBS electrode on fibre bundles projecting from or to the hypothalamic nuclei involved in the regulation of feeding behaviour and metabolism is a plausible hypothesis, although a recent study assessing the global function of the hypothalamus in patients with PD after DBS did not show any abnormalities of the hypothalamic–adrenal, hypothalamic–gonadal or hypothalamic–somatotropic axes.44 In patients with PD with STN DBS, despite high leptin concentrations secondary to the weight gain, one study reported an increase of the orexigenic NPY117 ,118 and hypothesised that DBS might make the hypothalamic neurons of the infundibular nucleus resistant to the anorexigenic effect of leptin. Normalisation of cortisol levels in PD has also been reported after DBS44 ,118 ,119 and its anabolic effect has been suggested as responsible for weight gain.43
Hypothalamic histopathological changes: neuronal populations in the lateral hypothalamic area (orexin and MCH neurons) are inhibited by leptin, activated by ghrelin and promote feeding (figure 2). As described previously, pathological studies have shown a severe reduction of both neuronal populations in patients with PD correlated with disease severity.29 ,30 A recent study also demonstrated the presence of Lewy body pathology involving the infundibular nucleus even at preclinical stages and these pathological changes increased with clinical progression.120 However, Lewy pathology did not show a correlation with the severity of weight loss suggesting that hypothalamic functional deficits rather than classical PD pathological changes may be responsible for the weight fluctuations.
Hedonic system: dysregulation of the dopaminergic mechanisms of hedonic control of feeding behaviour might also contribute to weight changes in PD. Although dopaminergic medications are generally reduced following STN DBS surgery, eating disorders secondary to behavioural changes following DBS may occur due to abnormalities of dopaminergic signalling similar to the alterations believed to be responsible for impulse control disorders.121 The involvement of hedonic dysregulation in the pathogenesis of PD weight gain is further supported by changes in metabolism after DBS in some of these brain areas including the orbitofrontal and anterior cingulate cortices using PET imaging.122
The mechanisms responsible for weight fluctuations in PD are far from understood but current evidence does not support the classic view of an energy intake/expenditure imbalance secondary to motor symptoms and complications of PD. Instead, these data suggest a disruption of hypothalamic mechanisms of feeding regulation with complex interactions with peripheral signals, hedonistic control mechanisms and other external factors (medication and DBS).
Therapeutic implications
Only a few studies have assessed nutritional interventions and there is insufficient evidence for specific recommendations. However, it is now well accepted that nutritional assessments should be part of the routine work-up of patients with PD and dietary intervention can improve the PD-related weight abnormalities. Individualised dietetic advice improves nutritional status and quality of life in malnourished patients with PD on medical treatment123 and nutritional intervention has been shown to be effective in weight control in patients with PD after DBS-STN surgery.124 Owing to the competing interaction for intestinal absorption between L-dopa and aminoacids, dietary interventions focusing on protein manipulation have been suggested in patients with PD on treatment with L-dopa and motor fluctuations. While there is insufficient evidence to support low-protein diets, they may induce weight loss and nutritional deficits in the long term. Protein redistribution interventions have shown an improvement in motor function with better results when carried out in early stages of the disease.125
Key points
Weight loss has been consistently reported in patients with PD. Those treated with DBS show a rapid and excessive weight gain.
This weight fluctuation is not explained by an energy expenditure/intake imbalance secondary to PD complications. Complex disruption of central (mainly hypothalamic) and peripheral mechanisms of feeding regulation may account for these weight fluctuations.
Nutritional advice is recommended in malnourished patients with PD and those with excessive weight gain after DBS. Protein redistribution interventions may improve motor control in patients with PD with motor fluctuations.
Osteoporosis and bone metabolism
Epidemiology
Patients with PD have an increased risk of fractures, most commonly affecting the hip.126 Subsequent clinical outcome tends to be poorer than the general population.127 A meta-analysis of nine studies showed a combined effect of the risk of fracture in patients with PD (OR) of 2.28 (95% CI 1.83 to 2.83).126 Indeed, PD has been found to be the strongest single comorbidity contributing to fracture risk in the Global Longitudinal Study of Osteoporosis in Women cohort.128
Pathophysiology and abnormalities in PD
A significant factor is the increased risk of falls inherent to PD (secondary to postural instability, gait freezing, orthostatic hypotension, motor fluctuations and cognitive impairment). In addition, patients with PD have abnormalities of bone metabolism which also contribute to the increased risk of fractures (figure 3). A meta-analysis confirmed significantly reduced bone mineral density at the femoral neck, lumbar spine, total hip and total body126 in PD compared with healthy controls. Using T-score values, the overall combined mean difference was significantly lower in patients with PD (−1.05; 95% CI −1.26 to −0.84).126 Immobility and reduced BMI, both commonly seen in PD, are risk factors for osteoporosis, but several other factors disrupting bone metabolism may contribute to bone loss.

{kind=link}
{kind=link}
{kind=link}
Bone health assessment and management in patients with PD. Factors influencing fracture risk in patients with PD and proposed assessment and management recommendations (see text). BMI, body mass index; DEXA, dual X-ray absorptiometry; FRAX, fracture risk assessment tool; OT, occupational therapy; PD, Parkinson's disease.
Role of vitamin D
Vitamin D has a crucial role in bone metabolism and deficiency results in bone loss by compensatory hyperparathyroidism. There is increased prevalence of vitamin D deficiency in patients with PD compared with healthy controls, as high as 55% of patients in some studies129 ,130 and patients with other neurodegenerative conditions.131 This suggests that this is an intrinsic feature of the disease and not just secondary to reduced sunlight exposure. Vitamin D has important effects on brain function and its receptors are expressed in dopaminergic neurons of the substantia nigra.132 It has been hypothesised that chronic vitamin D deficiency contributes to PD pathogenesis.133 The potential association of these two conditions is supported by the longitudinal study by Knekt et al134 showing that pre-existing vitamin D deficiency increased the risk of developing PD in a cohort of 3173 Finnish participants after adjustment for potential confounders. Patients with highest vitamin D concentration had a RR=0.33; 95% CI 0.14 to 0.80 of developing PD in comparison to the patients with the lowest concentration. A possible link at the transcriptional level has also been suggested, though studies looking for an association between some vitamin D receptor polymorphisms and the risk of PD have yielded conflicting results.135
Role of homocysteine
Hyperhomocysteinaemia is an independent risk factor for fractures through a dual mechanism reducing bone mineral density and disrupting cross-linking of collagen.136 Homocysteine has been shown to be elevated in L-dopa-treated patients with PD compared with controls, but similar results were not found in drug-naïve patients.137 Plasma concentration correlates with disease severity and patients with high concentrations have increased risk of hip fractures (RR=2.42; 95% CI 1.21 to 3.63).138 The underlying mechanisms causing hyperhomocysteinaemia in patients with PD are not understood, however L-dopa therapy and possibly vitamin B12 and folate deficiency may be involved.136 ,137
Therapeutic implications
Despite the substantial fracture risk associated with PD, bone health assessment and management have been largely ignored and no clinical guidelines address this issue specifically in patients with PD. Taking into account these limitations, several recommendations can be made (figure 3).
Fracture risk estimation: fracture risk assessment tool (FRAX) and Qfracture are useful tools to estimate fracture risk and guide those who should undergo dual X-ray absorptiometry (DEXA) for a more accurate evaluation of bone mineral density. FRAX assessment might be slightly superior in assessing patients with PD in the neurology clinic.139
Bisphosphonates: until evidence exists to support patients with PD having different DEXA thresholds for antiosteoporotic therapy, it seems reasonable to apply general population recommendations regarding treatment with bisphosphonates.140 Both risedronate and alendronate have demonstrated an improvement of bone mineral density and reduction of hip fractures in patients with PD.141–143
Vitamin D: levels should be routinely measured in patients with PD and replaced if deficient. Vitamin D supplementation144 and increased sunlight exposure145 have both demonstrated an amelioration of hypovitaminosis D, an increase in bone mineral density levels and a reduction in the fracture risk for patients with PD.
Non-pharmacological therapies: an integrated approach including non-pharmacological therapies such as exercise and lifestyle modifications should be included as part of a holistic care of PD.
Key points
Patients with PD have an increased risk of fractures and reduced bone mineral density.
In addition to factors inherent to the disease increasing the risk of falls, there is a disruption of bone metabolism including vitamin D deficiency and hyperhomocysteinaemia.
Routine bone health assessment and estimation of fracture risk is recommended with consideration of vitamin D supplementation if deficient, non-pharmacological therapies and treatment with bisphosphonates if indicated.
Conclusion
Metabolic and neuroendocrine abnormalities are common in PD and have important clinical implications. Clinicians should be aware of these abnormalities and include their assessment as part of routine clinical practice. Recognition and treatment of neuroendocrine and metabolic disturbances will intuitively improve PD care and patients' quality of life. The underlying pathophysiology of these disturbances warrants further research. A better understanding of their pathogenesis may lead to accurate peripheral biomarkers of these abnormalities, which in turn may enable the development of more effective targeted therapeutic interventions and neuroprotective drugs.
Acknowledgments
The authors would like to thank Áine Cassidy for her help in the preparation of the figures.
References
Footnotes
Contributors EDP-F wrote the first draft, contributed to project conception and organisation. DPB, PMB, TF and RAB revised and critically reviewed the manuscript for intellectual content. TTW contributed to project conception and organisation, and critically reviewed the manuscript for intellectual content.
Funding RAB is partly supported by an NIHR award of a Biomedical research Centre to Addenbrooke's Hospital/University of Cambridge. TTW receives funding from the Reta Lila Weston Trust for Medical Research.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.