Article Text

Abstract
Objective To evaluate variability and predictability of disability trajectories in moderately advanced and advanced multiple sclerosis (MS), and their modifiability with immunomodulatory therapy.
Methods The epochs between Expanded Disability Status Scale (EDSS) steps 3–6, 4–6 and 6–6.5 were analysed. Patients with relapse-onset MS and having reached 6-month confirmed baseline EDSS step (3/4/6) were identified in MSBase, a global observational MS cohort study. We used multivariable survival models to examine the impact of disease-modifying therapy, clinical and demographic factors on progression to the outcome EDSS step (6/6.5). Sensitivity analyses with varying outcome definitions and inclusion criteria were conducted.
Results For the EDSS 3–6, 4–6 and 6–6.5 epochs, 1560, 1504 and 1231 patients were identified, respectively. Disability trajectories showed large coefficients of variance prebaseline (0.92–1.11) and postbaseline (2.15–2.50), with no significant correlations. The probability of reaching the outcome step was not associated with prebaseline variables, but was increased by higher relapse rates during each epoch (HRs 1.58–3.07; p<0.001). A greater proportion of each epoch treated with higher efficacy therapies was associated with lower risk of reaching the outcome disability step (HRs 0.72–0.91 per 25%; p≤0.02). 3 sensitivity analyses confirmed these results.
Conclusions Disease progression during moderately advanced and advanced MS is highly variable and amnesic to prior disease activity. Lower relapse rates and greater time on higher efficacy immunomodulatory therapy after reaching EDSS steps 3, 4 and 6 are associated with a decreased risk of accumulating further disability. Highly effective immunomodulatory therapy ameliorates accumulation of disability in moderately advanced and advanced relapse-onset MS.
Statistics from Altmetric.com
Introduction
Whether currently available immunomodulatory therapies may modify disability trajectories in patients with moderately advanced and advanced multiple sclerosis (MS) remains to be answered.1 ,2
Three large cohort studies have explored factors affecting the disability accrual at various stages of MS.3–5 While many demographic and clinical features, including early relapse activity, age and sex, have been implicated in the accumulation of disability in early disease, these studies were largely unable to explain the variation in disease progression in moderately advanced MS (defined in these as the period between Expanded Disability Status Scale6 (EDSS) steps of 3 or 4, and 6). A small number of candidate predictors, such as early and late relapses,5 prior disease duration5 and sex,3 were inconsistent across these studies. It has therefore been suggested that disability accrual at later MS stages is primarily driven by neurodegeneration and is largely independent of inflammation.4 ,7
Prior studies did not assess the effects of immunomodulatory therapies, and their data sets preceded the use of novel and potentially more effective MS treatments, such as natalizumab,8 fingolimod,9 alemtuzumab,10 dimethyl fumarate11 and cladribine.12 Although these therapies are known to prevent relapses and reduce first disability progression events, their effect on long-term disability accumulation, especially in the less inflammatory stages of MS, remains an important question that is still to be addressed.1 ,13
The objectives of our study were to evaluate variability and predictability of disability trajectories in MS, and to explore whether disability accrual in moderately advanced and advanced MS is modifiable with immunomodulatory therapy.
Methods
Ethics statement
The MSBase cohort study14 (registered with the WHO ICTRP, ID ACTRN12605000455662) was approved by both the Melbourne Health Research Ethics Committee and local ethics committees in all participating centres (or exemptions granted as per local regulations). Where required, enrolled patients provided written informed consent.
Patient population and data collection
Longitudinal data from 32 336 patients from 108 MS centres in 32 countries were extracted from MSBase in December 2014. Data quality procedures were applied as described elsewhere,15 and only information from centres contributing ≥10 records to the MSBase cohort was used.
All data were recorded as part of routine clinical practice, with most centres practising near real-time data entry in relation to clinical visits. The MSBase protocol stipulates minimum annual updates of the data set, although patients with less frequent visits were included. The data entry portal was either the iMed patient record system or the MSBase online data entry system. Only prospectively acquired data were included in the analysis, with the exception of date of disease onset, which is typically determined retrospectively. Prospective follow-up for each patient was defined by the dates of the first and last EDSS entry.
Disability milestones and inclusion criteria
This study consisted of three separate analyses, each addressing a distinct MS epoch defined by different neurological disability. The primary analysis concerned the epoch between EDSS steps 3 (including step 3.5; moderate disability but unrestricted ambulation) and 6 (severe disability, unilateral assistance required to walk ≥100 m). EDSS steps 3 and 3.5 were combined as in previous studies;4 ,5 in addition, we have observed that the distributions of the prebaseline and postbaseline disability trajectories were consistent for EDSS steps 3 and 3.5 (data not shown). Two secondary analyses addressed the epochs between EDSS steps 4 (moderate disability and/or walking distance >500 m but not unrestricted) and 6, and between EDSS steps 6 and 6.5 (bilateral assistance required to walk ≥20 m). For each epoch (EDSS 3–6, 4–6 and 6–6.5), a separate population of patients with clinically definite relapse-onset MS were selected using the following inclusion criteria:
Patients must have reached the initial EDSS step of the respective epoch (3 or 3.5 for the 3–6 epoch, 4 for the 4–6 epoch, or 6 for the 6–6.5 epoch), confirmed over ≥6 months without any interval regression (confirmation EDSS scores recorded within 30 days of a preceding relapse were excluded); this was defined as the study baseline. Patients had ≥12 months of prospective follow-up prior to baseline, and at least two postbaseline visits ≥3 months apart. A minimum required data set consisted of year of birth, sex, date of the first clinical presentation of MS, disease course at onset, treatment and relapse information.
The EDSS 3–6 and 4–6 epochs were selected to emulate the natural history studies.3–5 The EDSS 6–6.5 epoch was chosen as the smallest measurable change in disability during advanced disease, in order to maximise sensitivity of our study to the accrual of clinically significant disability.
Study end points
The outcome of interest was the time from baseline to EDSS step 6 (for the EDSS 3–6 and 4–6 epochs) or 6.5 (for the EDSS 6–6.5 epoch), confirmed over ≥3 months with no interval regression (confirmation EDSS scores recorded within 30 days of a preceding relapse were excluded). The choice of confirmation time for baseline and end point EDSS score was based on greater stability of disability progression events at higher EDSS scores.16 Patients not attaining this outcome were censored at their last recorded EDSS score. Disability was scored by accredited scorers (online Neurostatus certification was required at each centre) using the EDSS.
The slopes of the EDSS trajectories prior to and following baseline were calculated with a linear regression over the prebaseline EDSS scores or the postbaseline EDSS scores (including the baseline score in both).
Clinical characteristics
Relapses were defined as the occurrence of new symptoms or exacerbation of existing symptoms persisting for ≥24 h, in the absence of concurrent illness or fever, and occurring ≥30 days after a previous relapse.17 Confirmation by increased EDSS was not required.
Annualised relapse rate and proportion of time on disease-modifying therapy were calculated for each of the three epochs and their respective prebaseline periods. The overall proportion of time on disease-modifying therapy, both prior to and during each epoch, was stratified according to the estimated higher efficacy therapies (mainly represented by natalizumab and fingolimod, but also including alemtuzumab, dimethyl fumarate, cladribine, rituximab and mitoxantrone) or lower efficacy therapies (mainly represented by interferon β preparations and glatiramer acetate, but also including teriflunomide).8–11 ,18 ,19 Time on therapy was defined by recorded starting and termination dates; for disease-modifying therapies where extended effects are recognised, estimated effect duration was used to calculate time on therapy: mitoxantrone (3 months from recorded treatment date), rituximab (6 months), alemtuzumab (5 years) and cladribine (12 months).
Statistical analysis
Statistical analysis was carried out by NL and TK using R V.3.1.0 (http://www.R-project.org). All hypotheses were tested at the two-tailed 0.05 level of statistical significance.
The variability in disease progression was examined through individual EDSS slopes for the prebaseline and postbaseline periods. Coefficient of variation was calculated as the ratio of slope SD and mean. For each period, Pearson's r was calculated to evaluate the correlation between prebaseline and postbaseline slopes.
Median times to confirmed EDSS step 6 (for the EDSS 3–6 and 4–6 epochs) or 6.5 (EDSS 6–6.5 epoch) were estimated. The associations between the demographic (sex, age at baseline) and clinical patient characteristics (MS duration at baseline, annualised relapse rate prebaseline and during the epoch, proportion of time on higher and lower efficacy therapies prebaseline and during the epoch, and rate of treatment initiations prebaseline) and the time to the outcome EDSS step were analysed with multivariable Cox proportional hazard models. These models were designed based on the results of a series of univariate Cox models and were adjusted for total duration of recorded prospective follow-up and, in women, the proportion of time pregnant. Where the assumption of the proportionality of hazards was violated as per statistical tests of Schoenfeld residuals, Weibull accelerated failure time models were applied instead. Continuous variables with a non-normal distribution were transformed using Box-Cox transformations.
Each of the primary and two secondary analyses was accompanied by three sensitivity analyses, where: (1) in addition to the definition provided above, the EDSS end point was required to be sustained without regression for the remainder of the available follow-up, (2) the inclusion criteria were altered to include patients reaching an EDSS step equal to or greater than the defined initial EDSS step for each epoch and (3) the analysis used a nested model taking into account patients' country of residence.
Role of the funding source
The study was conducted separately and apart from the guidance of the sponsors.
Results
Patients
Of the 32 336 patients in the MSBase cohort, the following number of patients with relapse-onset clinically definite MS fulfilled the inclusion criteria for the EDSS 3–6, 4–6 and 6–6.5 epochs: 1560, 1504 and 1231, respectively. These comprised 3415 unique patients. The majority of the excluded patients have not yet reached moderately advanced MS or had insufficient prebaseline follow-up. Of patients meeting the above criteria, 74–78% were included per epoch. Figure 1 details patient disposition information. The number of patients contributed per MSBase centre is provided in online supplementary table S2. Table 1 summarises demographic and clinical data for each epoch's cohort.
Characteristics of the study populations
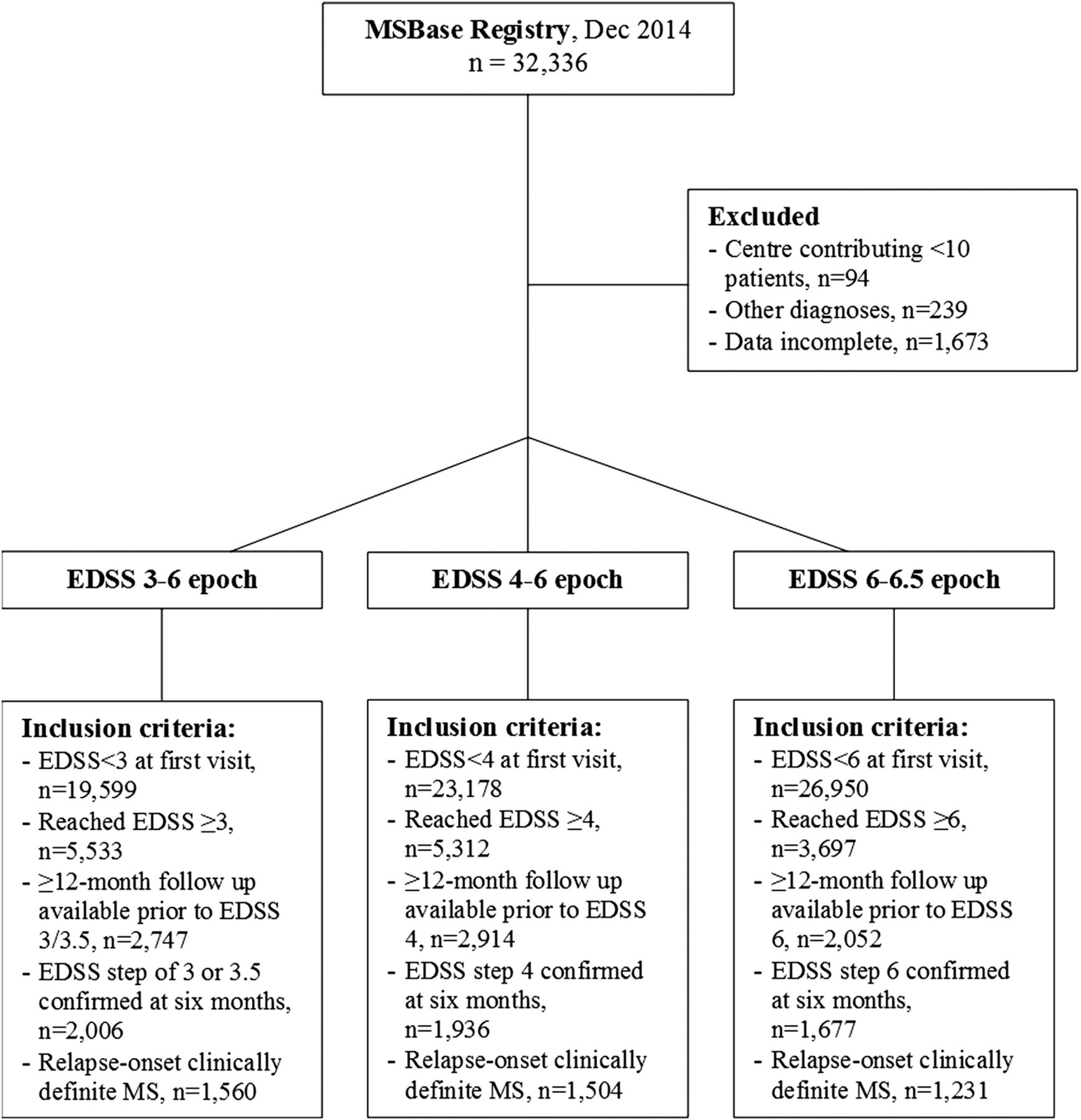
CONSORT diagram of patient disposition. EDSS, Expanded Disability Status Scale; MS, multiple sclerosis.
Supplemental material
Disability trajectories
Progression slopes (mean±SD) for the prebaseline period (EDSS 3/3.5: 0.34±0.38; EDSS 4: 0.38±0.39; EDSS 6: 0.56±0.51 EDSS steps/year) and for the postbaseline period (EDSS 3/3.5: 0.15±0.38; EDSS 4: 0.17±0.38; EDSS 6: 0.10±0.24 EDSS steps/year) were highly variable, as evidenced by large coefficients of variation (0.92–1.11 prebaseline and 2.15–2.50 postbaseline; figure 2A–C). No correlations were found between the prebaseline and postbaseline slopes (EDSS 3/3.5: r=0.01, p=0.57; EDSS 4: r=−0.001, p=0.97; EDSS 6: r=−0.03, p=0.37; figure 2D).

Disability trajectories pre-EDSS and post-EDSS steps 3, 4 and 6. EDSS trajectories between first recorded EDSS and baseline, and between baseline and last recorded EDSS for the three studied epochs: (A) baseline EDSS of 3 or 3.5, (B) baseline EDSS of 4 and (C) baseline EDSS of 6. (D) Scatterplot of prebaseline and postbaseline disability trajectory slopes. No correlations were found between prebaseline and postbaseline slopes. EDSS, Expanded Disability Status Scale.
Determinants of the progression of disability
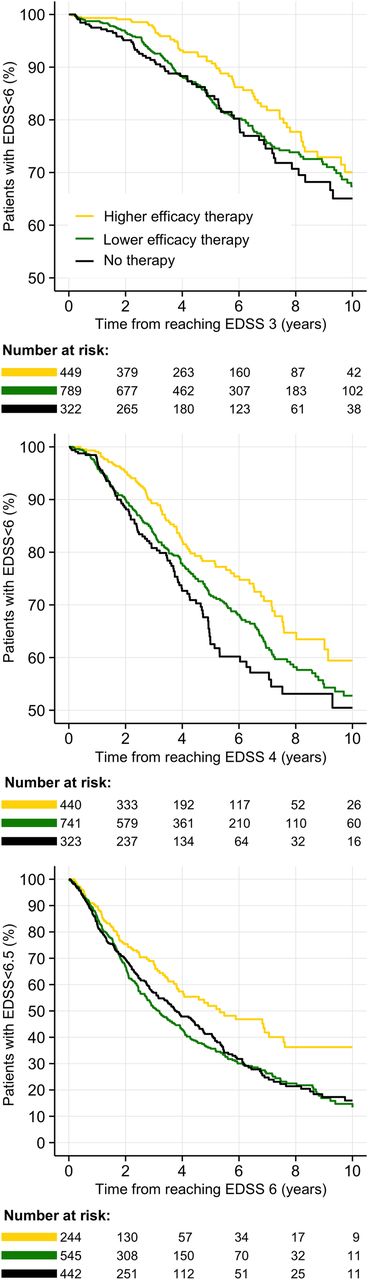
Results of multivariable survival models are shown in table 2. For all three epochs, higher annualised relapse rates during the epoch significantly increased the risk of reaching the EDSS end points (6 or 6.5), while an increasing proportion of the epoch spent on higher efficacy therapies significantly decreased this risk (illustrated in figure 3). For the primary analysis (EDSS 3–6 epoch), no prebaseline variables were associated with the probability of reaching EDSS step 6. For the EDSS 4–6 epoch, increased risk of reaching EDSS step 6 was associated with greater prebaseline exposure to higher efficacy therapies, male sex and shorter disease duration. For the EDSS 6–6.5 epoch, increased risk of reaching EDSS step 6.5 was associated with younger age at baseline, male sex and lower prebaseline relapse rate.
Determinants of progression to the confirmed outcome disability level
{kind=link}
{kind=link}
{kind=link}
Probability of reaching disability end points per epoch. Kaplan-Meier curves of the proportion of patients reaching disability end points during each epoch, stratified by exposure to therapy during the epoch. The strata reflect the highest efficacy of the administered therapy (here visualised as a categorical variable). Top: EDSS 3–6 epoch; middle: EDSS 4–6 epoch; bottom: EDSS 6–6.5 epoch. EDSS, Expanded Disability Status Scale.
For the EDSS 3–6, 4–6 and 6–6.5 epochs, median survival time to end points (years) was 17.3 (quartiles: 8.3–25.0), 11.4 (quartiles: 4.8–23.4) and 3.7 (quartiles: 1.7–7.2), respectively. The number (percentage) of patients reaching EDSS end points for each epoch was 296 (19%), 406 (27%) and 671 (55%), respectively.
Sensitivity analyses
Results of the sensitivity analyses are provided in online supplementary tables S3–S5. Taking into account the country of residence had no significant effects on the results of the primary analyses. The associations between exposure to higher efficacy therapies or the higher relapse rates during the studied epochs and the risk of attaining the EDSS end points were confirmed in full extent. The only exception was a lack of effect of annualised relapse rate within the sensitivity analysis including patients with EDSS step 6 or more at baseline for the EDSS 6–6.5 epoch. In addition, the sensitivity analyses including patients with EDSS step 3/4/6 or higher at baseline showed that patients with greater exposure to lower efficacy therapies during each epoch were less likely to reach the EDSS end points; however, this association was of smaller magnitude than that observed for higher efficacy therapies. For the EDSS 4–6 analysis, the effect of greater prebaseline time on higher efficacy therapy was supported by both sensitivity analyses, while neither demonstrated any effect of prebaseline disease duration. Finally, the sensitivity analyses reproduced some of the effects of male sex, older age at baseline and (for the EDSS 6–6.5 epoch) prebaseline relapse rate; however, these observations were inconsistent.
Discussion
Our study demonstrates that disability progression in moderately advanced and advanced MS is highly variable and, surprisingly, amnesic to prior disease activity. Features of early disease course, including relapses, disability trajectory, disease duration or treatment status, largely do not predict the rate of progression during later epochs. Contrastingly, we have found that once patients develop moderately severe and severe disability, lower relapse rates and greater persistence on highly effective immunomodulatory therapy significantly decrease the risk of further disability accrual. Together, this most likely represents an effect of immunomodulatory therapy on relapse-dependent disability progression. This effect of immunomodulation is independent on other factors, including prior disease activity and treatment.
Previous studies have identified factors associated with early disease progression.3–5 However, the ability to predict the course of more advanced MS has been limited and varied among these studies. These observations have led to a two-stage hypothesis, with the first stage representing a therapeutic window for modifying disease trajectory, which then becomes uniform in the second stage of disease.4 Our results concur that the disability trajectory in moderately advanced and advanced disease is independent of earlier disease characteristics, including previous disability trajectory, relapse activity or exposure to immunomodulatory therapy. As a milestone defining the two stages of MS course, EDSS step 3 was proposed,4 but Confavreux et al3 have reported a similar dichotomy between the epochs preceding and following EDSS steps 4 and 6. In fact, we have observed this dichotomy at various time points, including EDSS steps 3, 4 and 6. Amnesic disease trajectory therefore represents a more general MS characteristic, with clinical variables pertaining to any disease epoch affecting that epoch exclusively, with little effect on subsequent epochs.
In contrast to the study of Leray et al,4 we have shown that disability trajectories in moderately advanced MS are highly variable. Determinants of this variability, such as relapse rate, provide opportunities to modify disease course even at later disease stages. Our observation of the highly significant deleterious effect of greater relapse rates during each epoch is in contrast to some of the previous studies. While some studies only examined the effect of the presence/absence of relapses,4 ,20 Scalfari et al5 reported an unexpected association between a higher relapse count after the second year of disease and a reduced risk of disability progression. Unlike relapse rate, however, relapse count is confounded by time: patients with longer time to progression are exposed to a greater cumulative hazard of relapses. We have confirmed this assumption by substituting annualised relapse rate with relapse count in our models. We have noted a reversal in the polarity of HRs, creating an artefactual relationship similar to that reported by Scalfari et al (data not shown). Thus, the previously reported association between higher relapse count and lower probability of disability progression is a result of confounding. Using relapse rates, which are by definition time-adjusted and less susceptible to bias, we have demonstrated that greater relapse activity is associated with worsening of disability during moderately advanced MS, which is in keeping with a similar association demonstrated in earlier disease.21 ,22
While a large body of evidence indicates that immunomodulatory therapy reduces relapse rate, studies of the treatment effect on disability trajectories once significant disability has been attained are largely lacking.23–25 Our results demonstrate that sustained exposure to more effective immunomodulatory agents (here mainly represented by fingolimod and natalizumab) but not lower efficacy agents (here mainly represented by interferon β preparations and glatiramer acetate) mitigates further accumulation of disability even after significant disability has been attained (here quantified as EDSS step 3, 4 or 6). This observation is compatible with the outcomes of long-term follow-up extensions of randomised clinical trials in relapsing–remitting MS, which reported long-term benefits of early treatment with interferon β preparations or glatiramer acetate.23 ,26
A recent study has shown an association between the number of relapses and the hazard of reaching EDSS step 6 after the onset of progressive disease (in primary and secondary progressive MS).13 Interestingly, this study has also reported a decreased hazard of EDSS step 6 among patients who received immunomodulatory therapy during the progressive stage of disease. Together with our findings, these observations imply that even at the more advanced stages of MS, inflammation, which may manifest with relapses, contributes to the accumulation of permanent disability. In fact, Frischer et al27 showed that neurodegeneration in progressive MS is proportional to the magnitude of ongoing inflammatory activity. This concept has important therapeutic implications, as it justifies immunomodulatory therapy in patients with more advanced MS.
The observational character of our data represents the main limitation of the present study. However, evaluation of long-term disability trajectories and their response to therapy in a randomised trial is impractical and unethical,28 ,29 and all previous long-term follow-up studies in MS were based on observational cohorts. In order to minimise the impact of potential biases, we only used prospectively acquired data (mitigating recall bias), applied a rigorous data quality control procedure (reducing data entry errors, as described elsewhere15), defined a minimum required follow-up, adjusted the analyses for follow-up duration and used survival models with censoring (controlling attrition and selection biases), and modelled the outcomes in a series of two-step multivariable models adjusted for multiple potential confounders. Moreover, the independence of the main study outcomes (ie, the effects of relapse rate and therapy on disability accrual) from the definition of sustained disability accrual and the disability inclusion criteria was demonstrated by the sensitivity analyses. Our study was conducted in a large patient cohort representative of clinical practice at tertiary MS centres in multiple countries. This maximises the generalisability of our results given that treatment availability varies greatly across jurisdictions. In order to provide sufficient power to evaluate the effect of persistence on therapy, we grouped the available immunomodulatory therapies into two broad categories, based on the magnitude of their effects observed in randomised trials.8–12 ,18 ,19 Up to 30% of patients were exposed to higher efficacy disease-modifying therapies during the studied epochs. As a result, the distribution of the proportion of time on higher efficacy therapies was markedly right-skewed and its mean and median values were relatively low. We also acknowledge that the crude stratification according to the estimated treatment efficacy precludes any conclusions regarding the efficacy of individual treatments. However, rather than compare the effect of individual preparations, our aim was to explore the class effect of immunomodulation on the accumulation of disability in moderately advanced disease.
Contrasting the previous studies,4 our results suggest that disability accumulation in moderately advanced and advanced MS remains substantially driven by inflammatory activity. This hypothesis is supported by the observation that disability trajectories in moderately advanced and advanced relapse-onset MS are modifiable with immunomodulatory therapies. This observation, together with the general concept of the disease trajectory amnesic to the previous disease activity, leads us to conclude that prior disease activity should not preclude ongoing treatment, even when more advanced disability milestones have been reached (such as EDSS step 3, 4, or 6). While we demonstrate an under-recognised benefit of therapy in more advanced MS, this must nonetheless be weighed against the risks of individual immunomodulators in clinical decision-making. Our conclusion is highly relevant to the current debate resonating in the American MS community, concerning discontinuation of disease-modifying therapy in patients with MS.1 ,2 It also has important implications for the management of advanced disease, as well as treatment availability in jurisdictions where immunomodulatory therapies are only provided to patients with relatively mild disability.30
Acknowledgments
The authors wish to thank all patients and their carers who have participated in this study and who have contributed data to the MSBase cohort.
References
Footnotes
AL, RA, JL-S, MS and HB contributed equally.
Collaborators Contributing members of the MSBase Study Group are listed in online supplementary table S1.
Contributors NL conducted the analysis, and drafted and revised the manuscript. TK edited the manuscript. AL, RA, JL-S, MS, HB and TK conceptualised and designed the study, interpreted the analysis, and have revised and approved the manuscript. EH, DH, MT, GIz, PD, MG, AP, PG, RH, FGM, PS, EP, RB, CO-G, VVP, CRa, DS, GIu, CB, FG, JO, FV, CRo, EC, SF, SH, MPA, ND, VJ and TS contributed substantially to data acquisition, interpretation of the analysis, and have revised and approved the manuscript. NL and TK had full access to all the data in the study, conducted the analysis, and take responsibility for the integrity of the data and the accuracy of the analysis.
Funding This study was financially supported by National Health and Medical Research Council (practitioner fellowship 1080518, project grants 1083539 and 1032484 and centre for research excellence 1001216), and University of Melbourne Research Fellowship. The MSBase Foundation is a not-for-profit organisation that receives support from Merck, Biogen, Novartis, Bayer-Schering and Sanofi. The study was conducted separately and apart from the guidance of the sponsors.
Competing interests AL is a Bayer, Biogen, Genzyme, Merck Advisory Board Member. She received travel grants and honoraria from Bayer, Biogen, Merck, Novartis, Sanofi and Teva, research grants from Bayer, Biogen, Merck, Novartis, Sanofi and Teva, travel and research grants from the Associazione Italiana Sclerosi Multipla. RA received honororia from Biologix, Biogen, Bayer, Genpharm, Genzyme, Merck-Serono, GSK and Novartis, and served on advisory board for Biologix, Biogen, Bayer, Genpharm, Genzyme, Novartis, Genzyme, Merck-Serono and Novartis. JL-S has accepted travel compensation from Novartis, Biogen and Merck Serono. Her institution receives the honoraria for talks and advisory board commitment and also clinic support from Bayer Health Care, Biogen, CSL, Genzyme Sanofi, Merck Serono and Novartis. MS has participated in, but not received honoraria for, advisory board activity for Biogen, MerckSerono, BayerSchering, Sanofi Aventis and Novartis. EH received speaker honoraria and consultant fees from Biogen, Merck Serono, Novartis, Genzyme and Teva, as well as support for research activities from Biogen, Merck Serono and research grants from Charles University in Prague (PRVOUK-P26/LF1/4 and Czech Ministry of Health (NT13237-4/2012). DH received speaker honoraria and consulting fees from Biogen, Merck Serono, Teva and Novartis, as well as support for research activities from Biogen and research grants from Charles University in Prague (PRVOUK-P26/LF1/4) and Czech Ministry of Health (NT13237-4/2012). MT received speaker honoraria from Biogen-Idec, Bayer-Schering, Sanofi Aventis, Merck-Serono, Teva , Novartis and Almirall; has received research grants for her Institution from Biogen-Idec, Merck-Serono, and Novartis. GIz received speaking honoraria from Biogen, Novartis, Sanofi, Merck Serono and Teva. PD did not declare any competing interests. MG received consulting fees from Teva Canada Innovation, Biogen, Novartis and Genzyme Sanofi; lecture payments from Teva Canada Innovation, Novartis and EMD Serono. MG has also received a research grant from Canadian Institutes of Health Research. PG is a Novartis, Teva-neuroscience, Biogen and Genzyme advisory board member, consultant for Merck Serono, received payments for lectures by Merck Serono, Teva-Neuroscience and Canadian Multiple Sclerosis Society, and received grants for travel from Teva-Neuroscience and Novartis. RH received honoraria as consultant on scientific advisory boards from Merck-Serono, Biogen, Genzyme-Sanofi and Teva, research funding from Merck-Serono and Biogen, and speaker honoraria from Sanofi-Genzyme and Novartis. FGM received honoraria or research funding from Biogen, Genzyme, Novartis, Teva Neurosciences, Mitsubishi and ONO Pharmaceuticals. PS received travel grants and speaking honoraria from Bayer Schering, Biogen, Merck Serono, Novartis, Sanofi/Genzyme and Teva. EP served on scientific advisory boards for Genzyme and Biogen; he has received honoraria and travel grants from Sanofi Aventis, UCB, Lundbeck, Novartis, Bayer Schering, Biogen, Merck Serono, Genzyme and Teva; he has received travel grants from Associazione Marchigiana Sclerosi Multipla e altre malattie neurologiche. RB received speaker honoraria from Bayer Schering, Biogen, Genzyme, Merck Serono, Novartis, Sanofi-Aventis, Teva; research grants from Bayer Schering, Biogen, Merck Serono, Novartis, Sanofi-Aventis, Teva; congress and travel/accommodation expense compensations by Almirall, Bayer Schering, Biogen, Genzyme, Merck Serono, Novartis, Sanofi-Aventis, Teva. CO-G received honoraria as consultant on scientific advisory boards from Biogen, Bayer-Schering, Merck-Serono, Teva and Novartis; has participated in clinical trials/other research projects by Biogen, GSK, Teva and Novartis. VVP has served on advisory boards for Biogen, Novartis Pharma and Sanofi-Genzyme; has received travel grants and consultancy fees from Biogen, Bayer Schering, Sanofi Aventis, Merck Serono, Sanofi-Genzyme and Novartis Pharma; has received research grants from Bayer Schering. CRa received research funding, compensation for travel or speaker honoraria from Biogen, Novartis, Genzyme and Almirall. DS received honoraria as a consultant on scientific advisory boards by Bayer-Schering, Novartis and Sanofi-Aventis and compensation for travel from Novartis, Biogen, Sanofi Aventis, Teva and Merck-Serono. GIu had travel/accommodations/meeting expenses funded by Bayer Schering, Biogen, Merck Serono, Novartis, Sanofi Aventis, and Teva. CB received conference travel support from Biogen, Novartis, Bayer-Schering, Merck-Serono and Teva; has participated in clinical trials by Sanofi Aventis, Roche and Novartis. FG has served on scientific advisory boards for Biogen Idec, Novartis and Sanofi Aventis and received funding for travel and speaker honoraria from Biogen Idec, Merck Serono, and Almirall. JO serves on scientific advisory boards for Biogen, Genzyme and Novartis; has received speaker honoraria from Biogen, Bayer-Schering, Genzyme, Merck-Serono, Novartis and Teva and research grants from Biogen, Merck Serono, Novartis and Teva. FV is an advisory board member for Teva Biogen Merck Serono and Novartis. CRo has received speaker honoraria from Bayer Schering, Novartis and Biogen, congress and travel expense compensations from Biogen, Teva, Merck Serono and Bayer Schering. EC received honoraria as consultant on scientific advisory boards by Biogen, Bayer-Schering, Merck-Serono, Genzyme and Novartis; has participated in clinical trials/other research projects by Merck-Serono, Roche and Novartis. SF received research funding, speaker honoraria and compensation for travel from and served as a consultant on advisory board for Bayer-Schering, Teva, Biogen, Merck Serono, Genzyme and Novartis. SH received honoraria and consulting fees from Novartis, Bayer Schering and Sanofi, and travel grants from Novartis, Biogen Idec and Bayer Schering. MPA received honoraria as consultant on scientific advisory boards by Biogen, Bayer-Schering, Merck-Serono, Teva and Sanofi-Aventis; has received research grants by Biogen, Bayer-Schering, Merck-Serono, Teva and Novartis. ND received funding from Bayer, Merck Serono, Biogen, Genzyme and Novartis. VJ has received conference travel support from Novartis. TS received compensation for travel from Biogen. HB has served on scientific advisory boards for Biogen, Novartis and Sanofi-Aventis and has received conference travel support from Novartis, Biogen and Sanofi Aventis. He serves on steering committees for trials conducted by Biogen and Novartis, and has received research support from Merck Serono, Novartis and Biogen. TK has served on an advisory scientific board for Merck-Serono, has received conference travel support and speaker honoraria from Novartis, Biogen, Sanofi Aventis, Genzyme, Teva, BioCSL and Merck Serono and has received research support from Biogen.
Ethics approval Melbourne Health Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial commentary