Article Text

Abstract
Background Evidence for efficacy of disease-modifying drugs in multiple sclerosis (MS) comes from trials of short duration. We report results from a 16 y, retrospective follow-up of the pivotal interferon β-1b (IFNB-1b) study.
Methods The 372 trial patients were randomly assigned to placebo (n=123), IFNB-1b 50 μg (n=125) or IFNB-1b 250 μg (n=124) subcutaneously every other day for at least 2 y. Some remained randomised for up to 5 y but, subsequently, patients received treatment according to physicians' discretion. Patients were re-contacted and asked to participate. Efficacy related measures included MRI parameters, relapse rate, the Expanded Disability Status Scale, the Multiple Sclerosis Functional Composite Measure and conversion to secondary progressive MS.
Results Of the 88.2% (328/372) of patients who were identified, 69.9% (260/372) had available case report forms. No differences in outcome between original randomisation groups could be discerned using standard disability and MRI measures. However, mortality rates among patients originally treated with IFNB-1b were lower than in the original placebo group (18.3% (20/109) for placebo versus 8.3% (9/108) for IFNB-1b 50 μg and 5.4% (6/111) for IFNB-1b 250 μg).
Conclusions The original treatment assignment could not be shown to influence standard assessments of long-term efficacy. On-study behaviour of patients was influenced by factors that could not be controlled with the sacrifice of randomisation and blinding. Mortality was higher in patients originally assigned to placebo than those who had received IFNB-1b 50 μg or 250 μg. The dataset provides important resources to explore early predictors of long-term outcome.
- Multiple sclerosis
- disease-modifying drugs
- interferon β-1b
- long-term follow-up study
- mortality
Statistics from Altmetric.com
Introduction
Patients with multiple sclerosis (MS) commonly live 30 or 40 y after disease onset.1 Long-term outcomes determine the key social, medical and economic impact of the disease. However, it is not possible to quantitate or adequately assess the overall effect of disease-modifying drugs (DMDs) on disease course. Obstacles include the impossibility of maintaining blinding and randomisation and the problems in assessing patients who discontinue treatment.
Pivotal trials have shown benefits from DMDs in patients with clinically isolated syndromes at risk of developing MS2–4 and with relapsing-remitting multiple sclerosis (RRMS).5–14 However, these trials have been of relatively short duration and the long-term treatment benefit is less clear.
Interferon β-1b (Betaferon/Betaseron; IFNB-1b) was approved for treating patients with RRMS following a pivotal study.5 In this study, treatment with IFNB-1b 250 μg for 2 y reduced the clinical relapse rate by 34% compared to placebo (p=0.0001). Final analysis at 5 y demonstrated that the clinical relapse rates each year were one-third lower in patients treated with IFNB-1b 250 μg than in placebo-treated patients.6 It was feasible for patients to defer treatment for 5 y or more allowing for the inclusion of a group randomised to placebo at the time of this study. Following completion of the pivotal trial, patients were under regular medical care and, thus, free to receive IFNB-1b 250 μg or other treatment as they became available over time, such as IFNB-1a intramuscularly (im), glatiramer acetate (GA), mitoxantrone, IFNB-1a subcutaneously (sc), in the case of one patient, natalizumab. MS treatments were chosen by treating physicians according to conviction making interpretation of long-term outcomes more difficult.
In a concurrent analysis of 16 y follow-up data from the pivotal study of IFNB-1b, no important safety concerns were identified in those originally receiving active treatment. Other studies have also looked at long-term outcomes associated with DMD treatment but the observation periods have been shorter.4 15–20 The purpose of this current analysis was to explore whether differences in clinical outcome can be detected at 16 y follow-up between the originally randomised groups or patient subgroups from the pivotal IFNB-1b trial, subsequent use of other treatments or discontinuation of treatment notwithstanding.
Methods
Patients and study design
The design for this study and the basic methods are described in detail elsewhere.21 The original pivotal IFNB-1b study was conducted in 11 clinical centres in the United States and Canada.5 6 22 Patients who participated in the original trial (n=372) were re-contacted by their original clinical study centre between January 2005 and October 2005 and were asked to participate in this 16 y follow-up study (ClinicalTrials.gov Identifier: NCT00206635) .21 Those who agreed to participate were assessed during 1 day or for up to 3 days, if necessary. If, for health or personal reasons, patients chose not to participate in person, they could provide limited information via a telephone interview. Ethics approval for the follow-up study was obtained from the institutional review boards or independent ethical committees of the participating centres. All patients gave written informed consent.
Treatment
During the original study patients were randomly assigned to receive placebo (n=123), IFNB-1b 50 μg (n=125) or IFNB-1b 250 μg (n=124) sc every other day for 104 weeks.5 22 Patients were asked to continue for a further 12 months' extension phase of double-blind treatment and evaluations, with some remaining on the study for up to 5 y. Once IFNB-1b 250 μg was approved in October 1993, all remaining patients were offered the commercially available product (Betaferon/Betaseron).
No specific therapeutic regime was adhered to as part of the follow-up study. Many patients were on DMDs other than IFNB-1b during the course of the study. Information on the treatment history of individuals was collected systematically, although it was not always possible to determine the precise duration of treatment for some DMDs because of uncertainty about the start and stop dates of treatments prescribed. In these cases, the most conservative estimate of exposure was assumed using the earliest and latest dates that patients could be confirmed to be on treatment.
Primary observations
A large number of outcomes were assessed and recorded in this descriptive and hypothesis-generating study. Deaths and medication history were gathered. Efficacy and effectiveness related measures included the level of disability/function as determined by the Expanded Disability Status Scale (EDSS)23 and the Multiple Sclerosis Functional Composite Measure (MSFC).24 Conversion to secondary progressive MS (SPMS) and its timing were based on investigator opinion, and from observation and review of patient case report forms for worsening disability for at least 6 months not relapse-attributable. Time to EDSS level 6.0 (intermittent or unilateral ambulation assistance required) and relapse rates were obtained from retrospective data review. Careful assessment of baseline characteristics is extensively described elsewhere.21 Other outcomes included MRI measures and assessments of cognitive function, and will also be reported elsewhere.
Subgroup analysis
The long-term follow-up (LT) patient population, nearly all of whom had received IFNB-1b at some time during the past 16 y, was then divided into three predefined groups according to duration of exposure to IFNB-1b 250 μg. These groups were arbitrarily defined as (1) IFNB-1b 250 μg for <10% of the time, (2) IFNB-1b 250 μg for 10–79% of the time or (3) IFNB-1b 250 μg for ≥80% of the time. These divisions were intended to identify the group that had received high-dose IFNB-1b continuously from the beginning of the trial (≥80%), another that had not received IFNB-1b 250 μg during the pivotal trial and had very limited exposure to IFNB-1b 250 μg thereafter (IFNB-1b 250 μg treatment for <10% of the time) and a third group comprising all other individuals. Comparison of these three groups was planned to analyse the relation between IFNB-1b use and progression-related outcomes.
Composite outcome
Heterogeneity is a key problem for any long-term study in which patients are no longer randomised or treated uniformly. However, endpoints commonly used in MS studies (such as relapse rate, EDSS score and time to progression of disability), despite their intrinsic variability, become harder and more discrete over the long term.25 26 We used a composite measure, the ‘negative disability outcome measure’, in this study to encompass unambiguous adverse outcomes. This was reached when an individual had an EDSS ≥6.0 or had been diagnosed as having converted to SPMS.
Statistical analysis
Statistical analyses in this study are necessarily descriptive. For continuous data, mean, SD and median are provided. Categorical data are described in frequency tables displaying the actual count as well as percentages.
Baseline characteristics and clinical outcomes at LTF, including the negative physical disability outcome, are presented for the LTF population (table 1)21 in groups according to randomised treatment during the pivotal study (placebo, IFNB-1b 50 μg or IFNB-1b 250 μg) and subgroups according to IFNB-1b exposure. Proportions of patients reaching the negative disability outcome (and its components) are provided together with median times to event.
Baseline characteristics of the long-term follow-up patients as per their original treatment assignment and duration of IFNB-1b treatment
Numbers of all identified patients who died, including those whose exact date of death was unknown, were presented by treatment arm. Time to death from onset of disease was evaluated using the Kaplan-Meier method; p values from log-rank tests for comparisons versus placebo serve descriptive purposes. For eight patients (four from the placebo group and two in each IFNB-1b group), missing dates of death were assumed to be the date of LTF. Baseline characteristics and clinical outcomes are also provided for groups according to IFNB-1b exposure.
Results
Study population
All 11 original centres participated. Among original study participants, 328/372 (88.2%) were identified. Of the patients identified, 293/328 were alive (89.3%) and 35/328 were deceased (10.7%). Case report forms were available for only 7 of the 35 deceased patients and these were included in the analysis. A total of 40 identified participants declined to give consent for follow-up. In total, case report forms were available from 260/372 (69.9%) identified patients. A total of 260 patients had EDSS evaluations, 192 had MRI evaluations and 179 had cognitive assessments in English.
Baseline characteristics
The baseline characteristics of the LTF population (as originally randomised and according to exposure to IFNB-1b) were similar among the three study groups and were representative of the original trial population (table 1)21. Data from patient case report forms showed that 74/260 (28.5%) patients were taking IFNB-1b 250 μg within 30 days of consenting to participate in the follow-up.
Duration of treatment
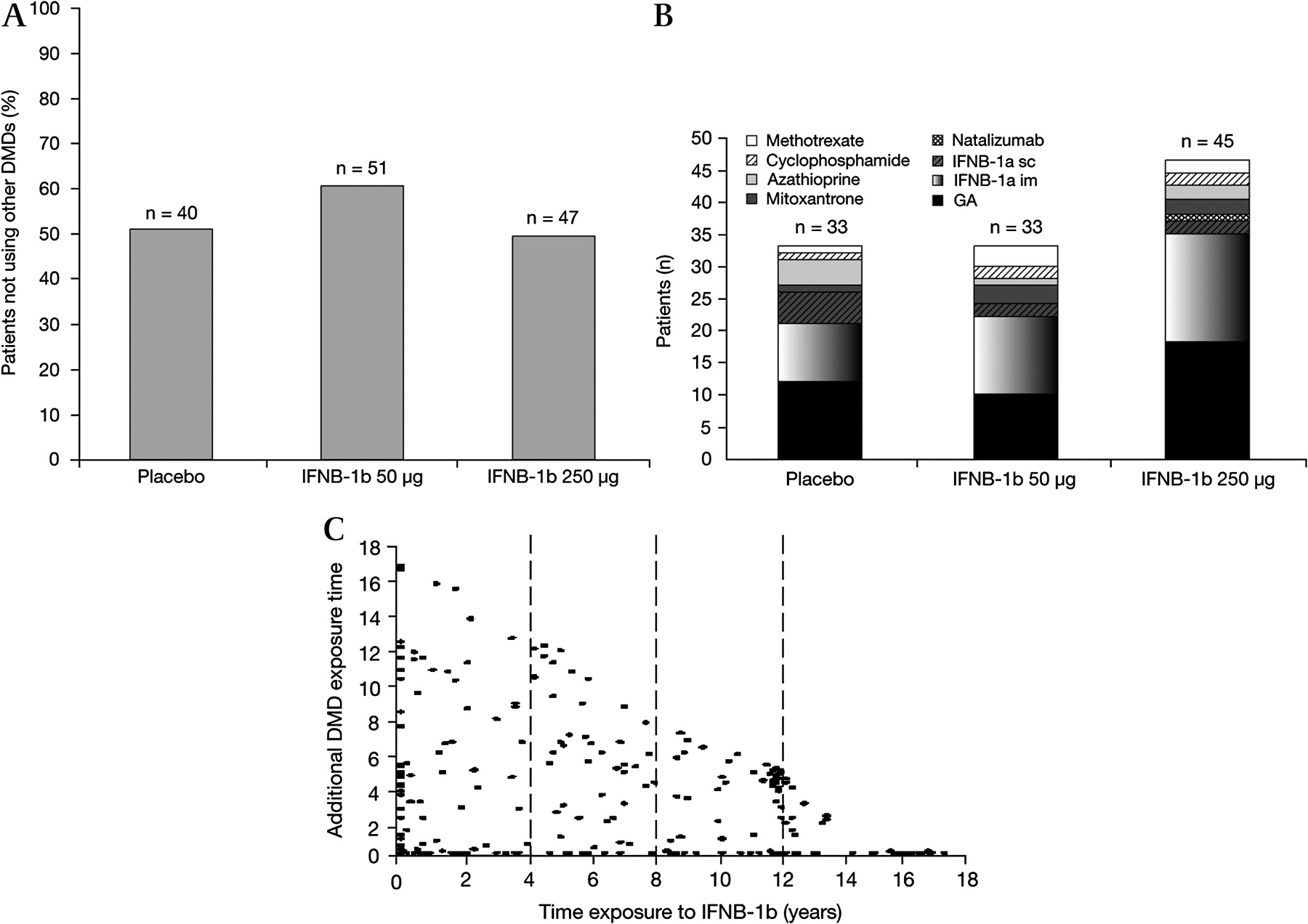
After completion of the pivotal trial, patients received treatments as recommended by their physician (mean 1.6 treatments per individual, SD=0.8). The majority (59.2%) received one MS treatment, 25.4% received two different MS treatments, 10.4% received three and 3.8% received four, although not necessarily continuously, for the LTF period (figure 1).
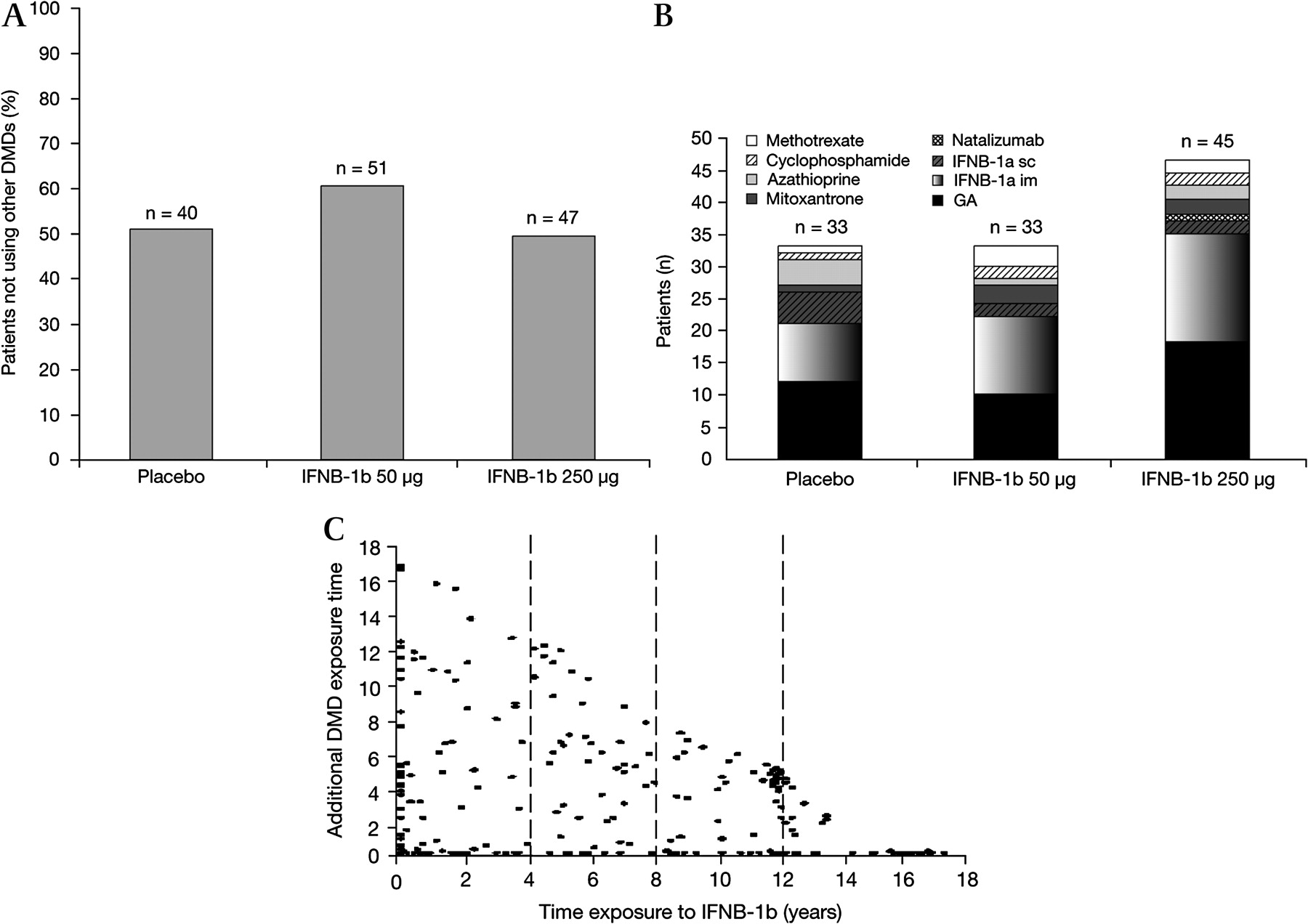
Patients using IFNB-1b only (A), disease-modifying drug (DMD) use other than IFNB-1b stratified by original treatment group (B) and additional DMD use versus duration of exposure to IFNB-1b 250 μg (C).
The ranges of time on any treatment varied considerably. Of the 260 patients studied, 40 (15.4%) received less than 6 months of MS treatment after completion of the clinical trial, whereas 28 (10.8%) remained on IFNB-1b treatment for >80% of the study period (>12.8 y). The majority of patients (85.8%) received IFNB-1b 250 μg at some time during the 16 y follow-up period. The median total length of exposure to IFNB-1b 250 μg since the start of the pivotal trial was 7.9 y. Overall, the duration of IFNB-1b exposure in the studied patients was 1784 patient-years versus 623 patient-years of exposure to other DMDs or immunosuppressive agents. There were fewer GA-treated patients and more azathioprine-treated patients in the placebo group than in the IFNB-1b-treated groups. In addition, there were generally similar proportions of IFNB-1a- and IFNB-1b-treated patients in the placebo and IFNB-1b-treated groups.
Mortality
In total, 35 deaths were recorded in the follow-up patients: 18.3% (20/109) of those identified from the original placebo group, 8.3% (9/108) of the original IFNB-1b 50 μg group and 5.4% (6/111) of the original IFNB-1b 250 μg group. Information on causes of death is available for nine patients and is reported elsewhere.21 Case report forms were only available for seven of the patients who had died. The lack of case report forms on the remaining 28 meant that these could not be included in the disability analyses.
The majority of deaths occurred >10 y from the start of the pivotal study and 20 y or more after onset of first symptoms (figure 2). Based on estimated survival rates from the start of the pivotal trial, patients evaluated from the 50 μg IFNB-1b and IFNB-1b 250 μg groups had a higher likelihood of survival than those randomised to placebo (p=0.0402 and p=0.0049, respectively (p values uncorrected)). In the current study, based on estimated survival rates, patients in the IFNB-1b 50 μg and IFNB-1b 250 μg groups appeared to have a better chance of survival than those randomised to placebo (p=0.0443 and p=0.0029, respectively). Treatment started, on average, 8 y after onset of MS symptoms.

Death by calendar year from onset of clinical symptoms.
Progression-related outcomes
Disability outcomes according to original randomisation showed that a total of 113 patients reached EDSS 6.0: 45.6% (36/79) of those originally assigned to placebo, 38.8% (33/85) of those assigned to IFNB-1b 50 μg and 45.8% (44/96) of those assigned to IFNB-1b 250 μg (table 2). The median times from onset of clinical symptoms to EDSS 6.0 for the original patient treatment groups were 14.5 y for placebo, 12.8 y for IFNB-1b 50 μg and 16.1 y for IFNB-1b 250 μg.
Disability outcomes at 16 y for patients participating in the long-term follow-up population according to the original pivotal trial treatment groups
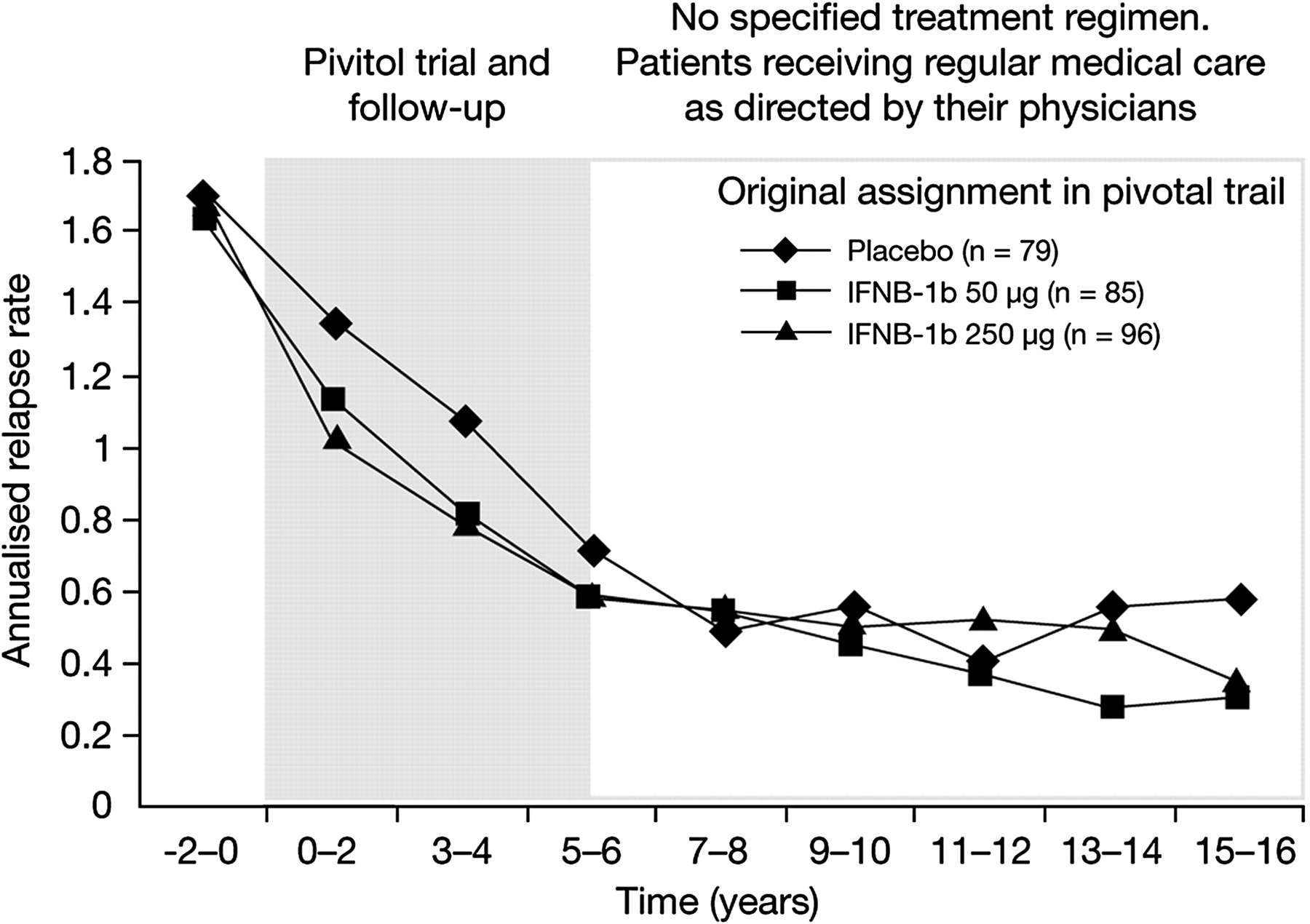
The pre-planned analysis of patients divided by <10% of the time on IFNB-1b (from entry into the pivotal study until the end of LTF duration), 10–79% of the time on IFNB-1b or ≥80% of the time on IFNB-1b gave unequal divisions (n=70, n=162 and n=28, respectively). Baseline characteristics are presented in table 1. While not statistically significant, the likelihood of reaching EDSS 6.0 was greater for the <10% IFNB-1b group (38.6%) and the 10–79% IFNB-1b group (46.9%) than for the ≥80% IFNB-1b group (35.7%). In addition, though also statistically insignificant, time from diagnosis to EDSS 6.0 was less for the <10% IFNB-1b group (8.3 y) than for the 10–79% IFNB-1b group (10.5 y) or for the ≥80% IFNB-1b group (13.6 y). Statistically insignificant differences were also observed regarding reduced incidence of SPMS (34.3% for the <10% IFNB-1b group, 44.4% for the 10–79% IFNB-1b group and 28.6% for the ≥80% IFNB-1b group) and increased time from diagnosis to SPMS (11.4 y for the <10% IFNB-1b group, 13.4 y for the 10–79% IFNB-1b group and 13.8 y for the ≥80% IFNB-1b group). Relapse rates prior to baseline, at baseline and in 2-yearly intervals on-study or post-study showed an overall decrease in annual relapse rate for all treatment groups from around 1.6–1.8 prior to baseline to approximately 0.3–0.6 at 15–16 y after initiating treatment (figure 3).
{kind=link}
{kind=link}
{kind=link}
Annualised long-term follow-up study relapse rate versus time from start of randomised study.
Composite outcome measure
Over half (55.7%) of the patients originally assigned to placebo reached the pre-defined negative physical disability outcome compared to 53.0% in the two combined IFNB-1b-treated groups and with 57.3% in the IFNB-1b 250 μg group. Composite outcomes according to treatment exposure are shown in table 3. One patient from the IFNB-1b 50 μg group died before reaching EDSS 6.0 or converting to SPMS, but was reported by a first cousin to have died of an MS-related cause and was, therefore, counted as having reached a negative outcome in the statistical analyses.
Time to the disability endpoints according to duration of IFNB-1b treatment
Discussion
Careful analysis of clinical data collected 16 y after initial randomisation of patients to the pivotal trial of IFNB-1b was carried out in the collected dataset. Long-term effectiveness of IFNB-1b was difficult to prove using traditional approaches, despite the nearly 90% patient ascertainment achieved in this study, as there was no parallel control group and assessment of efficacy is, at this stage, largely focused on the impact of the original treatment assignments. Non-traditional approaches to bias mitigation and data analysis will be considered in detail elsewhere. Mortality was reduced in patients originally treated with IFNB-1b versus placebo but the number of deceased patients in this study was small and it is not possible to confirm a survival benefit of IFNB-1b treatment. Such an effect will be reassessed in a planned 20 y follow-up. It is of course possible that IFNB-1B treatment could be somehow impacting on survival independent of any therapeutic action in MS.
Relapse rates were low in the 16-y trial population compared to baseline, but most of the patients received additional treatment following completion of the short-term study. This finding cannot be ascribed solely to use of IFNB-1b.
A study on the efficacy of GA concluded that patients who received GA continuously over 10 y during the follow-up period experienced better long-term outcome than patients who withdrew from treatment (EDSS increase of 0.50 vs 2.24).20 This 16 y follow-up study of patients treated with IFNB-1b found an apparent decrease in the incidence of reaching EDSS 6.0 or developing SPMS compared to those who discontinued treatment (>80% vs <10% exposure to IFNB-1b). However, patients who take a treatment continuously may be more likely to do so because of positive outcome resulting either from successful treatment or from less aggressive disease. Similarly, patients with poor outcome are likely to change treatments to seek greater clinical benefit. Therefore, patients continuing to receive treatment can be self-selected for positive outcome and these data do not necessarily imply a treatment effect.
Numerous factors confound interpretation of these data by original treatment assignment. This analysis necessarily sacrifices randomisation and blinding and lacks a true parallel control group. An additional confounder is incomplete identification and follow-up of all patients participating in the original trial. Such confounders have led to criticism of other long-term studies, such as the extension trial of the PRISMS study,4 9 which examined the benefits of up to 8 y of IFNB-1a treatment. It has been suggested that extension trials of this type support long-term safety more than long-term efficacy.27 These potential biases impose similar difficulty in the interpretation of the present efficacy data as analysed by traditional methods. However, our analyses of this 16 y follow-up have demonstrated support for the long-term safety of IFNB-1b.
An additional complication is the widespread self-selected or physician-selected use of other treatments, including methotrexate, cyclophosphamide, azathoprine, mitoxantrone, IFNB-1a im, IFNB-1a im sc and GA. Therefore, causality cannot be assigned to an outcome with complete confidence. There did not appear to be a bias when we examined the baseline characteristics and performance during the pivotal study of those participating in follow-up compared to those refusing and with those who could not be found, but differences in the latter group may have emerged during the ensuing years. Those patients who did not participate in a detailed follow-up study did less well during the randomised trial compared to those who participated in the LTF. There was no indication that the delay in treatment in the original placebo group versus the IFNB-1b-treated patients had an impact on disability. The probability of reaching EDSS 6.0 did not differ among the original treatment arms. The difference in start of any treatment between treated and placebo arms consisted of 2–4 y by which time patients were already into the second decade of disease. These results do not strongly bear on the value of ‘early treatment’.
This study provided the opportunity to focus on hard outcome measures that have face validity, such as EDSS ≥6.0, SPMS and death. There was little difference for EDSS 6.0 among the treatment arms but these data do not take into account the 28 deaths for which there were no case forms. The omission of these data may well obscure meaningful effects of treatment timing, because the death distribution was skewed towards an increased mortality in the placebo arm. If the deaths could be shown to be related to incremental disability, this would be more favourable to the treatment arms. The endpoints commonly used in short-term clinical trials, such as relapse rate and MRI outcomes, are indirect measures that, prior to this study, had an uncertain association with these long-term ‘hard’ outcomes or even shorter-term ones.28 The difference in mortality between the original patient groups is a novel observation that will be further explored.
The results here represent the longest available follow-up of any DMD and may also be the most complete and comprehensive. The final evaluation after 16 y from study entry comprises more than 4000 patient-years. This actually extends beyond two decades from disease onset on average, because, at study entry, mean duration of disease from clinical presentation was 8.02 y (SD=6.15). Other follow-up studies of DMD treatment have encountered similar difficulties with patient identification but have had shorter periods of observation.4 15–20 The duration of these trials may have been too short to identify a clear mortality benefit of treatment, which even this study can only propose. Life expectancy for patients with MS has been estimated to be between 5 and 10 years less than for individuals without the disease.29 30 In the Danish Multiple Sclerosis Registry, MS or complications of the disease accounted for more than half of the deaths that occurred.31 Whether or not DMD treatment can reduce the raised mortality risk for patients with MS is an important question and warrants further investigation.
Long-term data on the clinical outcomes of MS treatment are potentially of great importance to physicians, patients and third-party payers. However, conclusive evidence could not be gained using the methodologies used in short-term randomised clinical trials. Further exploration of methods for the interpretation and analysis of non-randomised long-term data is needed.
Acknowledgments
The authors wish to thank the team at PAREXEL MMS Europe for their support during the development of this manuscript, including: Mary Beth DeYoung and Rebecca Gardner for the preparation of the initial outline of this manuscript, which was prepared at a face-to-face meeting attended by all authors; David Morgan, for proof-reading and preparation of the figures, and Catherine Amey for collating revisions from co-authors. PAREXEL MMS Europe received payments from Bayer HealthCare Pharmaceuticals for this editorial support.
References
Supplementary materials
Lay Summary 81/8/907
Files in this Data Supplement:
Footnotes
Linked articles 208017.
16 y long-term follow-up study investigators: University of Chicago, Illinois, USA: B Arnason (Principal Investigator (PI)), A Reder, A Javed, A Noronha (Investigators (I]), B Harding-Clay, M Valentine, K Ivy (Study co-ordinators (SC)), J Fink, T Nader, M Lacy (Neuropsychological testers (NT)); University of California, San Francisco, USA: DS Goodin (PI), E Kornyeyeva (SC), D Cox (NT); Greenstein Associates & MS Institute, Philadelphia, USA: J Greenstein (PI), I Gold (SC); University of Maryland, Baltimore, USA: K Johnson (PI), C Bever, H Rus, R Shin (I), E Katz, V Wells, K Naunton (SC), M Rogerson (NT); Jefferson University Hospital, Philadelphia, USA: T Leist (PI), S Gallardo, J Davis (SC), A Okai (NT); University of Arizona, Tucson, USA: WA Sibley (PI), J Wendt, DA Weidman, RD Wachter (I), J Laguna, B Peterson (SC), D Brown (NT); University of Alabama at Birmingham, USA: K Bashir (PI), B Layton (SC), J Castillo, S Krzywanski (NT); Hôpital de Notre-Dame, Montréal: P Duquette (PI), R Dubois (SC), J Poirier (NT); Institut et Hôpital Neurologiques de Montréal, Canada: Y Lapierre (PI), S Hum (SC and NT); University of British Columbia, Vancouver, Canada: J Oger (PI), V Devonshire, J Hooge, A Traboulsee, P Smyth, S Hashimoto, L Kastrukoff (I), W Morrison, F Lum (SC), B Kosaka (NT); London Health Sciences Centre, Ontario, Canada: GPA Rice (PI), M Kremenchutzky, R Deshpande (I), J Lesaux (SC), H Armstrong (NT), J Moussa, T Bentall (Research assistants); University of Oxford, Oxford, UK: GC Ebers (Signatory Investigator), A Scalfari (I); University of London, London, UK: D Langdon (Neuropsychology Coordinator); Radcliffe Infirmary, Oxford, UK: J Gurd (Neuropsychology Coordinator); MRI Center, Vancouver, British Columbia, Canada: D Li, A Traboulsee (PI), R Tam (Research assistant), M Medina, A Riddlebough (SC).
Competing interests This study was sponsored by Bayer HealthCare Pharmaceuticals. At the time of the study, TB, CW, KB and AK were salaried employees of Bayer Schering Pharma AG, Berlin, Germany. GE has received research support from Bayer Schering Pharma AG/Bayer HealthCare Pharmaceuticals. AT has received honoraria from Bayer Schering Pharma AG/Bayer HealthCare Pharmaceuticals, Biogen, Serono and Teva. DL is Director of the UBC MS/MRI Research Group, which has been contracted to perform central analysis of MRI scans for therapeutic trials with Angiotech, Bayer HealthCare, Berlex-Schering, Bio-MS, Centocor, Daiichi Sankyo, Hoffmann-LaRoche, Merck-Serono, Schering-Plough, Teva Neurosciences, Sanofi-Aventis and Transition Therapeutics. DL has received research support from Bayer Schering Pharma AG/Bayer HealthCare Pharmaceuticals and honoraria from Bayer Schering Pharma AG/Bayer HealthCare Pharmaceuticals, Serono Symposia, Merck Serono, Biogen and Hoffman La Roche. DG has received research support and honoraria from Bayer Schering Pharma AG/Bayer HealthCare Pharmaceuticals. AR has received research support from the Berlex, Bayer, Biogen, Teva, Serono, National Institutes of Health, the National MS Society, the Brain Research Foundation, the American Academy of Allergy & Immunology, Howard Hughes Foundation, North American Symptomatic Carotid Endarterectomy Trial, Egypt Arab Republic Peace Fellowship, Turkish Ministry of Defense Fellowship Award and the State of Illinois, USA.
Ethics approval Obtained from the institutional review boards (IRBs) or independent ethics committees of the participating centres before LTF planning, which began in 2004.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial commentary