Article Text

Abstract
Background The homogeneous genotype and stereotyped phenotype of a unique familial form of amyotrophic lateral sclerosis (ALS) (patients homozygous for aspartate-to-alanine mutations in codon 90 (homD90A) superoxide dismutase 1) provides an ideal model for studying genotype/phenotype interactions and pathological features compared with heterogeneous apparently sporadic ALS. The authors aimed to use diffusion tensor tractography to quantify and compare changes in the intracerebral corticospinal tracts of patients with both forms of ALS, building on previous work using whole-brain voxelwise group analysis.
Method 21 sporadic ALS patients, seven homD90A patients and 20 healthy controls underwent 1.5 T diffusion tensor MRI. Patients were assessed using ‘upper motor neuron burden,’ El Escorial and ALSFR-R scales. The intracranial corticospinal tract was assessed using diffusion tensor tractography measures of fractional anisotropy (FA), mean diffusivity, and radial and axial diffusivity obtained from its entire length.
Results Corticospinal tract FA was reduced in sporadic ALS patients compared with both homD90A ALS patients and controls. The diffusion measures in sporadic ALS patients were consistent with anterograde (Wallerian) degeneration of the corticospinal tracts. In sporadic ALS, corticospinal tract FA was related to clinical measures. Despite a similar degree of clinical upper motor neuron dysfunction and disability in homD90A ALS patients compared with sporadic ALS, there were no abnormalities in corticospinal tract diffusion measures compared with controls.
Conclusions Diffusion tensor tractography has shown axonal degeneration within the intracerebral portion of the corticospinal tract in sporadic ALS patients, but not those with a homogeneous form of familial ALS. This suggests significant genotypic influences on the phenotype of ALS and may provide clues to slower progression of disease in homD90A patients.
- Amyotrophic lateral sclerosis
- diffusion tensor tractography
- corticospinal tract
- fractional anisotropy
- mean diffusivityALSimage analysisMRI
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/2.0/ and http://creativecommons.org/licenses/by-nc/2.0/legalcode.
Statistics from Altmetric.com
- Amyotrophic lateral sclerosis
- diffusion tensor tractography
- corticospinal tract
- fractional anisotropy
- mean diffusivityALSimage analysisMRI
Introduction
Amyotrophic lateral sclerosis (ALS) is a complex progressive neurodegenerative disease affecting upper (UMN) and lower (LMN) motor neurons of the motor cortex, brainstem and spinal cord.1 An estimated 90% of ALS cases are apparently sporadic. Clinical manifestations include muscle weakness, wasting and spasticity, resulting in death 2–5 years after onset. The remaining 10% of ALS cases are understood to be familial, with 20% of these being closely associated with over 100 predominantly missense genetic mutations2 in a gene that codes for cytosolic Cu/Zn binding superoxide dismutase (SOD-1) on chromosome 21.3 SOD-1 mutations are typically inherited in a dominant fashion, but recessive patterns of inheritance have been shown in patients homozygous for aspartate-to-alanine mutations in codon 90 (homD90A), representing the most common SOD-1 mutation.4 Although homD90A patients have a disease phenotype similar to sporadic ALS (see Weber et al5 for a review), they have a longer mean survival (mean 12.6 years vs 4 years), the reason for which is unknown.
Imaging techniques such as diffusion tensor MRI (DTI) have been used to quantify corticospinal tract degeneration in vivo in sporadic ALS patients.6–8 DTI can be used to estimate organisational properties of tissue microstructure and orientation of white-matter fibre tracts based on the principles of water displacement.9 Diffusion of water molecules is higher parallel to fibre bundles than perpendicular to them, creating a directional bias, which can be detected using DTI.10 Four principal outcome measures can be computed: fractional anisotropy (FA), a measure of directionality of diffusion, mean diffusivity (MD), a measure of overall diffusion of water in a voxel, axial diffusivity, which reflects diffusion along fibre bundles, and radial diffusivity, which reflects diffusion perpendicular to fibre bundles. By determining the relationship of diffusion information between adjacent voxels, detailed 3-D images of fibre tracts can be constructed (tractography) for quantitative analysis.11
The corticospinal tracts (CSTs) contain the major efferent UMN fibre projection connecting the motor cortex with the brainstem and spinal cord.12 Projections from UMN, originating in the motor cortex, converge in the corona radiata, descend through the posterior limb of the internal capsule and continue into the cerebral peduncles and pons before entering the spinal cord.13 By placing regions of interest (ROI) at several of these known anatomical locations, it is possible to delineate the CSTs and quantify UMN degeneration throughout their entire intracerebral length as reflected by abnormal diffusion values.
Previously, we have demonstrated differences between sALS, homD90A and controls using whole-brain voxelwise group analysis of FA.14 The current study used a different and newer analysis technique, diffusion tensor tractography to examine diffusion characteristics along the intracerebral corticospinal tract in the same cohort of patients with sporadic ALS and those homozygous for the D90A SOD-1 mutation. Using diffusion tensor tractography one can determine axial and radial diffusivity which cannot be measured using whole-brain voxelwise analysis approaches and provide information, which we hoped would allow us to understand better the pathophysiology of ALS in vivo. It was hypothesised that CST damage would be more pronounced in both ALS groups compared with age-matched healthy controls, but less extensively in homD90A compared with sporadic ALS patients. This is in line with previous comparative studies showing relatively preserved cortical inhibition,15 and more frontally dominant patterns of reduced cortical binding of [11C]-flumazenil,15 and volumetric cortical loss16 in homD90A ALS. In addition, we hypothesised that diffusion tensor tractography measures might correlate with clinical measures of disease severity, UMN pathology and disease duration.
Method
Subjects
Twenty-one patients with sporadic ALS (six female), seven with homozygous D90A SOD-1 ALS (six female) and 20 healthy control subjects (eight female) took part in the study. All but one of the subjects was right-handed, with no previous history of cerebrovascular disease, diabetes mellitus or hypertension. Patients with sporadic ALS were recruited from the King's Motor Neuron Disease Care and Research Centre, London, and diagnosed following clinical examination by a consultant neurologist. Other disorders were excluded by appropriate blood tests, neurophysiology and neuroimaging.1 Patients with sporadic ALS were classified according to the revised El Escorial criteria17 as four ‘definite,’ eight ‘probable’ and eight ‘possible.’ HomD90A ALS patients were recruited from Umeå University Hospital, Umeå, Sweden and travelled to London to take part in the study. Healthy volunteers with no history of neurological symptoms were recruited from the spouses and family members of patients and from members of local voluntary organisations. The study was approved by the King's College London, Institute of Psychiatry local research ethics committee and conformed to the standards set by the 1964 Declaration of Helsinki. Written, informed consent was given by all subjects prior to participation in the study.
Clinical evaluation
Clinical evaluations were performed on the day of MRI scanning. Disease duration was calculated for all patients in months from the time of onset. Patients were assessed by a neurologist (VW) using standardised disease rating scales. Global disease severity was assessed using the revised ALS functional rating scale (ALSFRS-R).18 The Ashworth Spasticity Scale19 was used in all patients to provide a specific measure of UMN dysfunction. In addition, patients underwent a more detailed assessment of UMN ‘burden’ by totalling the number of pathological UMN signs on examination.20 These were taken as subjectively pathologically brisk facial and jaw jerks; biceps, triceps, brachioradialis, knee and ankle reflexes; Hoffman's sign; and extensor plantar responses (all scored bilaterally) giving a maximum score of 16.
Data acquisition
Images were acquired from the whole brain using a 1.5 T GE Signa LX NV/i system (General Electric, Wakashua), incorporating actively shielded magnetic field gradients (maximum amplitude 40 mT/m) and a standard quadrature birdcage head coil. Images were acquired parallel to the anterior commissure–posterior commissure (AC-PC) line.
Using a multislice, peripherally gated EPI pulse sequence, each DTI volume was acquired from 60 contiguous 2.5 mm thick slices with field of view (FOV) of 240×240 mm and matrix size of 96×96, zero-filled to 128×128, ultimately giving an in-plane voxel size of 1.875×1.875 mm2. The echo time (TE) was 107 ms, with effective repetition times (TR) of 15 R–R intervals. Seven images were obtained at each location without diffusion weighting, with 64 images with a weighting of 1300 s/mm2 applied along directions uniformly distributed in space.21 Cardiac gated DTI data acquisition led to variations in scanning time in relation to each patient's pulse rate, and were on average 25 min.
Data preprocessing
Data processing was performed using the ExploreDTI v4.4 software package.22 The DTI data sets were corrected for head motion and eddy-current-induced geometric distortions. During this correction procedure, the b-matrix was adjusted for potential subject motion to ensure correct diffusion tensor estimates.23 The diffusion tensor was then calculated on a voxel-by-voxel basis using a weighted multivariate linear regression after logarithmic transformation of the signal intensities,24 and tensor matrix diagonalised to compute the eigenvalues and eigenvectors.
Image analysis
Data from one patient with sporadic ALS were not included in subsequent analyses owing to poor quality. Fibre tracking was performed with a standard DTI-based deterministic streamline fibre tracking method using all brain voxels with FA >0.2 in a seed region (described below) as a starting-point.25 Streamlines were propagated using a Euler integration method with a step size of 0.5 mm as described by Basser et al.25 Streamlines were stopped when FA was lower than 0.2, or the angle between two steps was greater than 35°. This acts to terminate tract trajectories that do not match natural white-matter pathways of the brain.26 Diffusion tensor tractography can be used to delineate fibre pathways by placing multiple ROI functions (SEED, AND and NOT) on anatomical landmarks. In order to determine the location of the corticospinal tract, detailed white-matter atlases were used to identify anatomical structures known to be associated with it.13 A neurologist experienced with white-matter-tract anatomy and tractography (MC) assisted with identification of the corticospinal tracts in order to ensure the accurate dissection of tracts during analysis. Qualitative assessment of images and identification of anatomical features were performed blind to subject status, as was fibre tracking. The corticospinal tract was primarily identified using directionally encoded colour maps27 to indicate the directionality of projection fibres via colour representations on axial slices (illustrated in figure 1A). ROIs were defined in each hemisphere using FA maps (illustrated in figure 1B). In the current study, a ‘SEED’ ROI was placed in the posterior limb of the internal capsule (illustrated in figure 1C) and an ‘AND’ ROI on the most caudal aspect of the cerebral peduncle (illustrated in figure 1D). The SEED and AND regions are chosen to be large enough to include all voxels that might be part of the CST at the relevant levels. Spurious fibres that were not associated with the corticospinal tract were eliminated by rejecting streamlines shorter than 20 mm or greater than 500 mm in length and also using a ‘NOT’ ROI, with deletion confirmed by a recalculation of tracts. The entire intracranial portions of the corticospinal tracts of both cerebral hemispheres (illustrated in figure 1E) were then separately analysed.

MR images and regions of interest used in the study. (A) Anatomical structures were located using directionally encoded colour maps. These maps were used to determine the most rostral component of the internal capsule and the most caudal aspect of the cerebral peduncle. (B) Grey-scale fractional anisotropy images. (C) Example of regions of interest (ROI) and tract configuration in 3-D. (D) ‘SEED’ ROI on the posterior limb of the internal capsule. (E) ‘AND’ ROI on the cerebral peduncle.
To provide further measures of diffusion, eigenvalues were sorted in order of decreasing magnitude (λ1>λ2>λ3). Diffusion parallel to fibre tract orientation is represented by the principal eigenvalue, λ1, giving a measure of axial diffusivity. Diffusion eigenvalues perpendicular to the main fibre tract (λ2 and λ3) were averaged and are referred to here as radial diffusivity.
Statistical analysis
A one-way repeated-measures ANOVA was applied to examine group differences in FA, MD, axial and radial diffusivity, with group as the between-subject factor and diffusion measures as the repeated measures within-subject factor. Appropriate post hoc analyses were performed on all significant group results. Correlation analyses were performed between diffusion measures (FA and MD) and clinical scores (disease duration, ALSFRS-R, Ashworth Spasticity Scale and UMN burden) for sporadic ALS and hom D90A patient groups separately using Pearson's correlation coefficient. Sensitivity, specificity and positive predictive values for mean CST FA were calculated for the sALS and control groups.
Results
Subject characteristics
Demographic and clinical data for all three groups are given in table 1. There was no significant difference in age between the three groups (p=0.62). The homD90A ALS patient group had a higher proportion of female subjects than sporadic ALS or controls. There was no significant difference in the global measure of disease severity (ALSFRS-R) between the two patient groups (p=0.74). There was no significant difference between sporadic ALS and homD90A patients in the UMN burden score (p=0.08). Data for the Ashworth spasticity scale were unavailable for one patient with sporadic ALS. The patient groups differed on the Ashworth Spasticity Scale with the homD90A patients more severely affected (p<0.001).
Demographic and clinical data of control subjects and patient groups
Fractional anisotropy and mean diffusivity
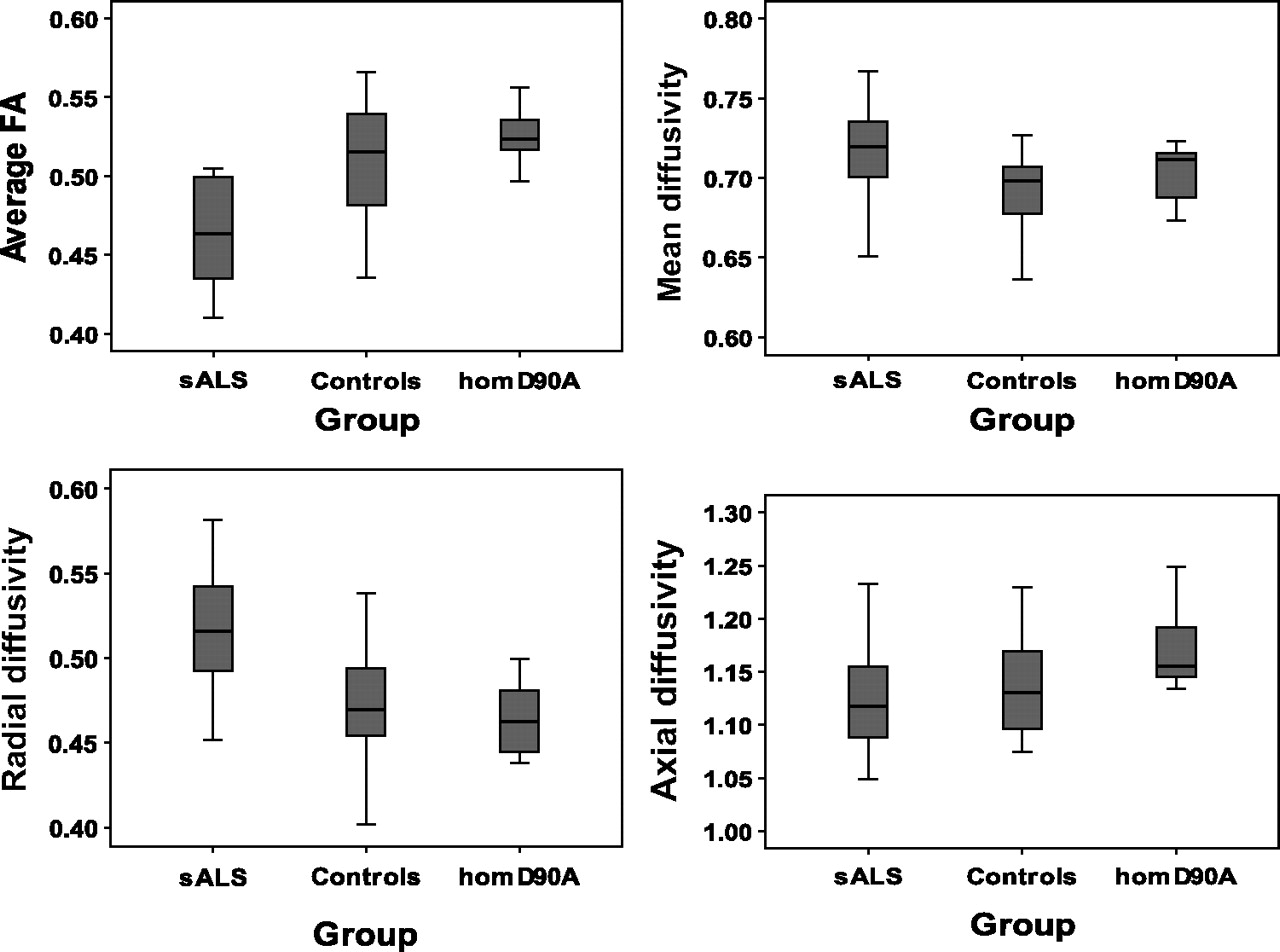
FA and MD results are summarised in table 2 and displayed in figure 2. Average values from both CSTs are presented. A one-way independent ANOVA revealed a significant group effect for measures of FA (p<0.001) but not MD (p<0.06) along the corticospinal tract. Post hoc tests revealed that FA was significantly lower in the CST in patients with sporadic ALS compared with both homD90A ALS patients (p<0.001) and controls (p<0.001). There were no differences in FA measures between homD90A ALS patients and controls. Using average FA values of the right and left CST gave sensitivity, specificity and positive predictive values of 85%, 63% and 72% respectively for discriminating sALS patients from controls. This is below a level that would be considered useful clinically.
Mean (SD) diffusion measures from the corticospinal tracts in patient groups and controls

Corticospinal tract fractional anisotropy (FA), mean diffusivity (×10−3 mm2/s), axial diffusivity (×10−3 mm2/s) and radial diffusivity (×10−3 mm2/s) in patients with sporadic amyotrophic lateral sclerosis (sALS), patients homozygous for aspartate-to-alanine mutations in codon 90 (D90A) and control subjects.
Radial and axial diffusivity
One-way ANOVA revealed a significant effect of group for radial (p<0.001), but not axial (p=0.16) diffusivity within the CST. Therefore, post hoc tests were carried out for radial diffusivity alone. These revealed that radial diffusivity in the CST was significantly increased in patients with sporadic ALS compared with both homD90A ALS patients (p=0.001) and controls (p<0.001).
Clinicoradiological correlations
Clinicoradiological correlations were examined for the sporadic ALS and homD90A ALS patient groups separately. There were no significant correlations between disease duration and any of the diffusion measures in either of the patient groups.
In the sporadic ALS group, a moderate negative correlation was noted between CST FA and the UMN burden score (r=−0.49, p=0.03) and Ashworth Spasticity Scales (r=−0.51, p=0.02) as illustrated by figure 3. However, there were no correlations between diffusion measures and the global measure of disease severity (ALSFRS-R), or between MD, radial diffusivity or axial diffusivity and clinical scores.

{kind=link}
{kind=link}
{kind=link}
Correlations between corticospinal tract fractional anisotropy and upper motor neuron (UMN) burden score (r=−0.49, p=0.03) and Ashworth Spasticity Scale (r=−0.51, p=0.02) in the sporadic amyotrophic lateral sclerosis group.
In the homD90A ALS group, there were no correlations between any of the diffusion measures (FA, MD, radial diffusivity, axial diffusivity) and the clinical scores.
Discussion
In this study, we examined genotype/phenotype interactions in ALS by quantifying differences in diffusion measures reflecting axonal damage in the intracerebral portion of the CST in familial and sporadic ALS patients. Despite the smaller size of the homozygous D90A SOD1 ALS group, the power to detect between-group differences remains good because of their homogeneity, as illustrated by figure 2. FA in the CST was markedly reduced in patients with sporadic ALS compared with familial ALS patients homozygous for the D90A SOD1 gene mutation. HomD90A ALS patients showed no differences to the control group on any of the diffusion measures. This is consistent with axonal degeneration within the intracerebral portion of the CST in sporadic ALS, but with apparent relative preservation in homD90A ALS patients, despite similar levels of upper motor neuron dysfunction and overall disability clinically. In addition, in sporadic ALS patients, reductions in CST FA were related to greater ‘bedside’ scores of UMN dysfunction, whereas in homD90A ALS patients there was no relationship between clinical UMN dysfunction and any of the intracerebral CST diffusion measures.
There was disparity in disease duration between the patient groups, with homD90A ALS patients having a longer disease duration compared with patients with sporadic ALS. This is consistent with the natural history of ALS in patients with the homD90A SOD1 gene mutation, where the disease is slowly progressive. We do not think that we are simply comparing rapidly progressing ALS (sporadic ALS) with slowly progressing cases that are in the early stages (homD90A) because measures of global (ALSFRS-R) and specific (‘UMN burden’) clinical severity are comparable. This implies that we are quantifying the pathology at a similar point in the disease process.
Patients were included in the study as ‘sporadic ALS’ if they had no family history of ALS. However, recent genetic studies have shown that 1–7% of apparently sporadic ALS patients carry a SOD1 gene mutation and in rare cases may carry TDP43, Angiogenin, FUS and Dynactin mutations. We did not perform genetic tests on the patients with apparently sporadic ALS, recruited into this study. While there is a theoretical possibility that the sporadic ALS group may have included patients carrying a gene mutation, it is extremely unlikely that this would be the rare homD90A SOD1 genotype.
Using diffusion tensor tractography, we have quantified differences in involvement of a key cerebral region (CST) between homD90A and sporadic ALS patients, that were suggested using whole-brain voxel-based analysis techniques of structural MRI and DTI. When these groups were both compared with controls, patients with sporadic ALS showed more pronounced areas of grey-matter atrophy within motor and premotor cortices than the familial group.16 FA was higher in motor and extra-motor regions in homD90A ALS patients compared with sporadic ALS (in addition to occipito-temporal and occipito-parietal areas), suggesting preservation of white-matter structure in these regions.14
To understand more about the possible differences in pathological mechanisms between homD90A ALS and sporadic ALS groups, we examined individual diffusion components that determine FA and MD (radial and axial diffusivity). This was not possible using our previous whole-brain FA voxelwise group-analysis approach.14 In patients with sporadic ALS, although there was a marked reduction in FA compared with the control group, MD was relatively preserved along the CST. This is a generally consistent finding in studies using DTI to examine axonal degeneration within the CST in sporadic ALS.7 28–31
In patients with sporadic ALS, we demonstrated that radial diffusivity (the degree of diffusion perpendicular to the white-matter tract) was significantly increased compared with controls, whereas axial diffusivity (the degree of diffusion parallel to the white-matter tract) did not differ from controls. The same profile of diffusion values has also been demonstrated in the corticospinal tract of sporadic ALS patients using an ROI approach31 and more recently with tractography.32 33 This suggests that reductions in FA in sporadic ALS are due to increased diffusivity of water molecules across white-matter fibres within the CST, rather than impedence of diffusivity along the tract. This pattern of diffusion values (reduced FA, preserved MD, increased radial but not axial diffusivity) is proposed to be highly suggestive of secondary damage to white matter, where there is anterograde (or Wallerian) axonal degeneration following a neuronal insult.34 This contrasts with primary damage to white matter, as occurs in strokes or multiple sclerosis, where there is a global increase in diffusivity of water molecules and a consistent, more marked increase in MD.34 In sporadic ALS patients, the profile of diffusion values consistent with Wallerian degeneration is in keeping with the notion that the predominant pathological insult in ALS is to cortical neurons. As suggested by animal models of Wallerian degeneration, it is likely that demyelination and an increase in isotropic tissue structures such as gliosis and extracellular matrix must be present in sporadic ALS patients (in addition to axonal loss) to produce this profile of diffusion values.35 36
In homD90A ALS patients, all of the diffusion components within the intracerebral portion of the CST were similar to those of healthy control subjects, despite levels of disability and clinical UMN involvement comparable with the sporadic ALS patients. The DTI findings indicate that there may be less Wallerian degeneration of the intracerebral portion of the CST in homD90A ALS patients compared with those with sporadic ALS, in keeping with the notion that there is relative preservation of the motor cortex in this patient group. A possible mechanism for the relative sparing of intracerebral motor regions in homD90A patients might be preservation of the activity of cortical inhibitory circuits. This is supported by the relative conservation of cortical inhibition determined neurophysiologically in homD90A versus sporadic ALS patients,15 and the differential reductions in [11-C]-flumazenil cortical binding.37
Similar to this study, in sporadic ALS patients a tight link has been previously shown between clinical UMN involvement and CST FA.29 38 39 Although we did not examine the profile of diffusion measures from the spinal cord in this study, we speculate that degeneration of the CST within the spinal cord, perhaps owing to a ‘dying-back’ process from the anterior horn,40 may instead account for the marked UMN dysfunction seen clinically in homD90A ALS patients.
In this study, we have quantified genotypic influences on the phenotype in ALS, with relative preservation of the intracerebral portion of the CST in ALS patients homozygous for the D90A SOD-1 gene mutation. Further investigation of possible protective processes within the cerebral cortex in homD90A ALS patients may provide clues to mechanisms which are responsible for their prolonged survival. Future studies could benefit from the higher signal-to-noise ratios and resolution available with 3 T and higher-field-strength scanners, as well as multicentre pooling of such rare groups.
Acknowledgments
We would like to thank all of the participants in the study for volunteering their time.
References
Footnotes
See Editorial commentary, p 827
Linked article 241521.
CRVB and KB are joint first authors.
Funding This study was supported by funding from the Motor Neuron Disease Association and the NIHR Biomedical Research Centre for Mental Health at South London and Maudsley NHS Foundation Trust and the Institute of Psychiatry, King's College London. Part of this work was funded through a personal Fellowship from the Wellcome Trust awarded to MRT in 2001. MRT is currently supported by the Medical Research Council/Motor Neurone Disease Association UK Lady Edith Wolfson Clinician Scientist Fellowship. PMA was supported by the Swedish Brain Power Consortium, the Swedish Brain Research Foundation, the Hållstens Research Foundation and the Swedish association for the neurologically disabled.
Competing interests None.
Ethics approval Ethics approval was provided by the Institute of Psychiatry, King's College London ethics board.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial commentary