Article Text

Abstract
Neuromyelitis optica (NMO) has been described as a disease clinically characterised by severe optic neuritis (ON) and transverse myelitis (TM). Other features of NMO include female preponderance, longitudinally extensive spinal cord lesions (>3 vertebral segments), and absence of oligoclonal IgG bands . In spite of these differences from multiple sclerosis (MS), the relationship between NMO and MS has long been controversial. However, since the discovery of NMO-IgG or aquaporin-4 (AQP4) antibody (AQP4-antibody), an NMO-specific autoantibody to AQP4, the dominant water channel in the central nervous system densely expressed on end-feet of astrocytes, unique clinical features, MRI and other laboratory findings in NMO have been clarified further. AQP4-antibody is now the most important laboratory finding for the diagnosis of NMO. Apart from NMO, some patients with recurrent ON or recurrent longitudinally extensive myelitis alone are also often positive for AQP4-antibody. Moreover, studies of AQP4-antibody-positive patients have revealed that brain lesions are not uncommon in NMO, and some patterns appear to be unique to NMO. Thus, the spectrum of NMO is wider than mere ON and TM. Pathological analyses of autopsied cases strongly suggest that unlike MS, astrocytic damage is the primary pathology in NMO, and experimental studies confirm the pathogenicity of AQP4-antibody. Importantly, therapeutic outcomes of some immunological treatments are different between NMO and MS, making early differential diagnosis of these two disorders crucial. We provide an overview of the epidemiology, clinical and neuroimaging features, immunopathology and therapy of NMO and NMO spectrum disorders.
- Neuroimmunology
- Multiple Sclerosis
- Myelopathy
- Neuroradiology
- Neuroophthalmology
Statistics from Altmetric.com
Introduction
Neuromyelitis optica (NMO) is an autoimmune inflammatory disorder with predilection for the optic nerves and spinal cord. Historically, neurologists, especially in Asian countries thought NMO to be a subtype of multiple sclerosis (MS), whereas others considered them distinct conditions.1 Many recent advances, in particular, the discovery of NMO-IgG, an NMO-specific autoantibody directed against aquaporin-4 (AQP4), the major water channel in the central nervous system (CNS), clearly identified NMO as a separate disease from MS.1 The term, NMO spectrum disorders (NMOSD), corresponds to restricted forms of the disorder which include recurrent optic neuritis (ON), relapsing transverse myelitis (TM), and some encephalitic presentations, which are common in children, but may herald the onset of the disease in patients of all ages. NMOSD are unified by the detection in serum or cerebrospinal fluid (CSF) of NMO-IgG (AQP4)-antibody).1
Herein, we review epidemiological, clinical, laboratory, diagnostic, pathological, pathophysiological and therapeutic data pertaining to NMO and NMOSD.
Epidemiology
There are very few epidemiological studies in NMO, but case reports and series have been reported from many countries across the continents. In two population-based studies in the Caribbean region, the prevalence of NMO was 2.5/105 in Martinique (French West Indies) and 0.52/105 in Cuba, and the incidence was 0.1/105 in the first study.2 ,3 A recent Danish study estimated a higher prevalence of 4.4/105.4 Beyond the prevalence in each region, another important epidemiological aspect to be considered is the differences in the regional distribution of NMO and MS worldwide. In a study in Thailand, there was a high proportion of AQP4-antibody-positive patients (39%) among the local cohort of idiopathic demyelinating diseases of the CNS if compared with European and American cohorts (less than 10%).5 Moreover, some AQP4 antibody-positive patients fulfilled the diagnostic criteria of MS, and thus, additional caution may be required when evaluating such patients.5 The onset age of NMO is commonly around the fourth decade of life, but the first attack may occur at any age from early childhood to elderly patients. The female predominance in NMO is observed in many published cohorts, with a female to male ratio ranging from 3:1 in France to 10:1 in Japan.6 ,7 The female predominance in NMO is usually higher than the one observed in MS, especially in AQP-4 antibody positive patients,8 but the reasons for such a high female predominance in NMO are not known.
Although NMO cases are usually sporadic, familial NMO has been reported in 3% of some cohorts.9 In a large cohort (177 NMO sporadic cases, including 14 NMO familial cases and 1363 matched controls) genotyping of 8 AQP4 single nucleotide polymorphisms (SNPs) of interest identified only one uncommon SNP, providing no evidence that genetic variation in AQP4 accounts for susceptibility to NMO.10 HLA and other immune-related genes could be associated with genetic susceptibility to NMO, but the details remain unknown.
Clinical features
NMO is clinically characterised by acute, severe episodes of ON and TM. Longitudinal follow-up studies revealed that the majority of patients have a relapsing disease course. NMO-IgG seropositivity, in particular, is predictive of a relapsing course. The radiological and laboratory evaluations usually reveal a lack of brain MRI lesions resembling MS, longitudinally extensive TM of more than three vertebral segments (LETM), low frequency of oligoclonal IgG bands, and high rates of AQP4-antibody positivity. Early studies identified patients with NMO as those with disease exclusive to the optic nerves and spinal cord. Subsequent extensive clinical experience with NMO-IgG-seropositive patients revealed that while NMOSD have a predilection for the optic nerves and spinal cord, 60% of patients develop brain MRI abnormalities11 including lesions localised in AQP4-rich periventricular regions.11 This led to revised diagnostic criteria for NMO, which no longer excluded patients with brain involvement from a diagnosis of NMO, and also incorporated NMO-IgG seropositivity as a supporting criterion.12 NMOSD include limited forms of NMO (recurrent ON or TM), brainstem disorders (including intractable hiccup and nausea,13 or vomiting,14 and hypothalamic disorders (including syndrome of inappropriate antidiuretic hormone secretion (SIADH))15 which may herald the onset of NMO. In these patients, the absence of the opticospinal phenotype or existence of other clinical features may pose additional diagnostic challenges, and thus, NMO-IgG seropositivity is critical to distinguish them from other diseases, including MS.
NMO patients with TM attacks usually present with LETM and severe clinical disability, demonstrated by para/tetraparesis, and sphincter disturbances. Disability in NMO is usually more severe than in MS, and is usually related to the severity of NMO relapses. In NMO, a secondary progressive phase, as seen in MS patients, is uncommon;16 and most disabilities arise from the often devastating discrete acute attacks. Fatigue and pain in NMO are common and affect a broader body area than those in MS, and significantly impact the patients’ quality of life.17 ON may present as a unilateral or bilateral event, depending on the affected region of the optic nerve and chiasm. Visual impairment is commonly severe in NMO occasionally with poor recovery.18 In a study of NMO and MS patients with ON, the overall retinal nerve fibre layer thickness (RNFLT) measured by optical coherence tomography was thinner in NMO than in MS, suggesting that ON attacks are more severe in NMO, and RNFLT was thinner in NMO patients with more relapses.19
MRI findings
MRI of affected optic nerve and spinal cord demonstrates swelling and loss of blood-brain barrier integrity. While the spinal cord lesions in MS tend to be shorter (less than one vertebral segment in length), smaller and peripherally located, the cord lesions in NMO are typically located centrally and, as already discussed, extend over three or more contiguous vertebral segments, and sometimes span the entire spinal cord (figure 1). On the other hand, brain MRI lesions in NMO are often silent clinically, and many are non-specific or indistinguishable from MS lesions.11 However, some MRI-observed brain abnormalities appear to be specific to NMO. These abnormalities may parallel midline AQP4-rich regions, particularly the hypothalamus and periaqueductal brainstem regions surrounding the ventricular system, and may also extend into the cerebral white matter (WM) and cerebellum.11 Other brain MRI findings reported in NMO include: solitary large tumefactive parenchymal and callosal lesions,11 ‘cloud-like’ enhancement,11 radial hemispheric lesions, linear brainstem lesions extending into the spinal cord, and posterior reversible encephalopathy syndrome (PRES)-like lesions. Large lesions may appear oedematous in the acute phase, with contrast enhancement and diffusion restriction. In the chronic stage, these lesions usually shrink and occasionally disappear.
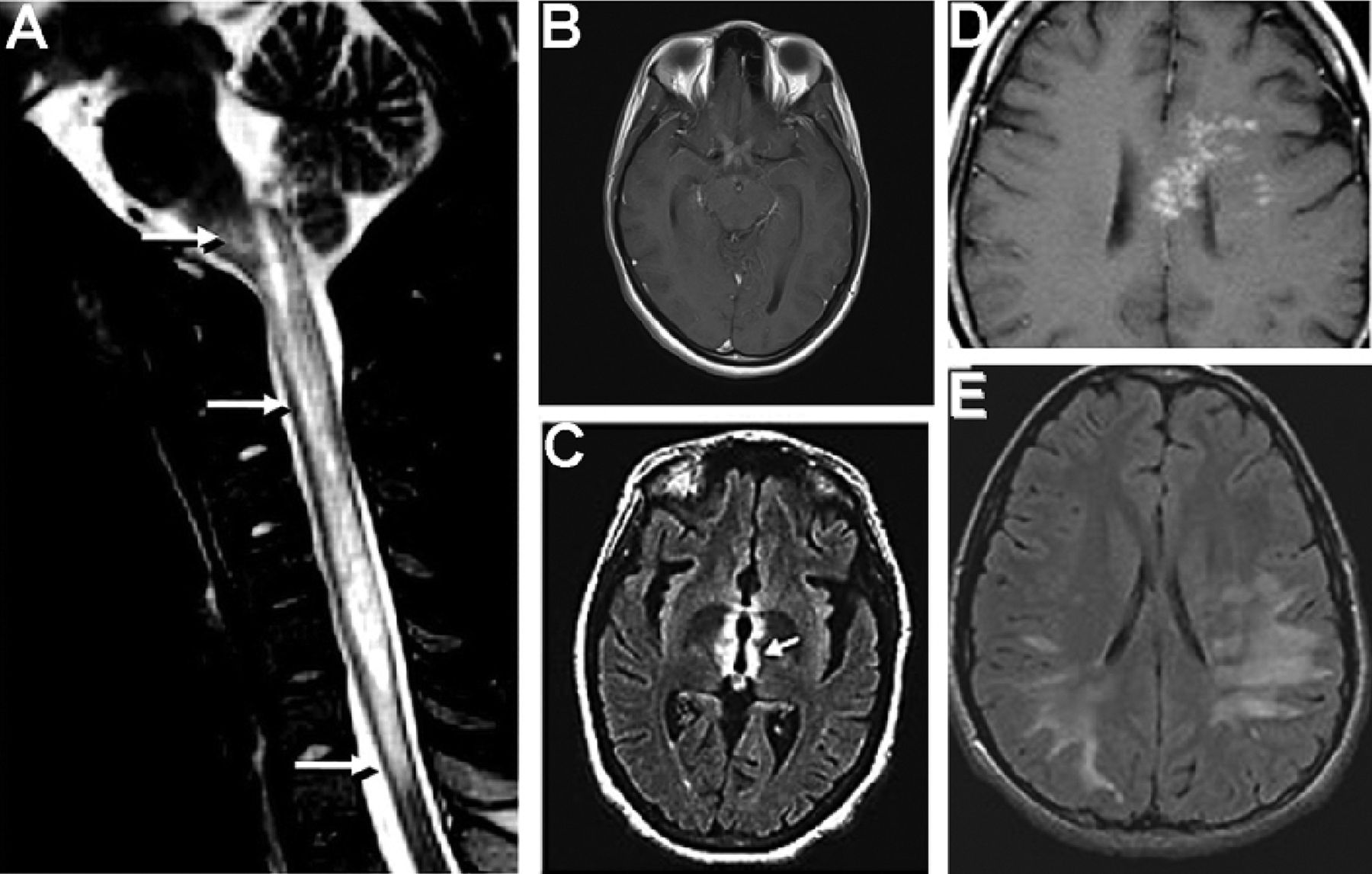
Radiological findings in neuromyelitis optica (NMO). (A) T2 saggittal MRI of spinal cord in a patient with myelitis and intractable vomiting demonstrates a longitudinal lesion extending from the upper thoracic cord to the medulla (arrows). (B) T1 axial MRI, postadministration of gadolinium in a patient with bilateral optic neuritis demonstrates enhancement of bilateral optic nerves and optic chiasm. (C) T2 fuid attenuation recovery (FLAIR) axial MRI of brain demonstrates signal abnormality (arrow) in the aquaporin-4-rich peri-IIIrd-ventricular diencephalon. (D) T1 axial MRI, postadministration of gadolinium demonstrates multiple areas of patchy enhancement with blurred margin in the adjacent regions (‘cloud-like enhancement’). (E) T2 FLAIR axial MRI demonstrates diffuse cerebral white matter abnormalities suspected in a boy initially suspected to have acute demyelinating encephalomyelitis (ADEM), but who ultimately had a relapsing course, and was found to be NMO-IgG positive. Reproduced with permission from Lippincott Willliams 2008 (A and B); American Medical Association 2006 and 2008, (C and E). Wiley publishers, 2009 (D).
Brain lesions are commonly observed in children; 68% of NMO-IgG seropositive children in one series; among them, 45% had brain symptoms corresponding to the MRI abnormalities.20 The MRI abnormalities encountered included clinically silent lesions limited to the periventricular regions; hypothalamic, thalamic and diencephalic lesions; long spindle or radial WM signal changes extending from the lateral ventricles into the cerebrum (unlike the short, pericallosal-confined Dawson's fingers of MS); WM abnormalities either extensive and confluent, or discrete and juxtaposed to or extending from the lateral ventricles; and brainstem and cerebellar lesions adjacent to the aqueduct of Sylvius and fourth ventricle. Endocrinopathies and disorders of water balance, which are recognised associations of NMO were attributable to hypothalamic involvement. The finding of hydrocephalus documented in one patient had an unknown relationship to AQP4 autoimmunity.20 Cerebral MRI lesions in affected children are sometimes large and extensive, resembling lesions of acute disseminated encephalomyelitis.20 As seen in adults, the MRI findings may be specific to presentations of intractable vomiting (linear area postrema and nucleus tractus solitarius medullary lesions) and SIAD (hypothalamic and periaqueductal lesions).13–15
As distinct from MS, MR spectroscopy parameters of measurement (ratios of N-acetylaspartate to creatine, choline to creatine and the absolute concentrations of the metabolites) have been reported to be normal in brains of patients with NMO, both those who have normal appearing grey and WM on conventional MRI, and those with brain lesions.21
NMO-IgG (AQP4-antibody) and other autoantibody findings
In 2004, Lennon and colleagues reported NMO-IgG, which has a characteristic tissue-binding pattern on indirect immunofluorescence with mouse brain slices (figure 2).22 This was detected in patients with NMO, and those with limited forms of NMO (such as LETM alone), and distinguished such patients from those with classical MS.22 The target antigen of NMO-IgG was subsequently confirmed to be AQP4, the predominant water channel in the CNS.23 This bidirectional channel is highly expressed in astrocytic foot processes in the regions critical for water, glutamate and potassium transport: the blood-brain barrier; synapses and paranodes adjacent to the nodes of Ranvier.24 The high specificity of NMO-IgG for the relapsing form of NMOSD (recurrent (and often bilateral) ON, recurrent (and usually longitudinally extensive) myelitis and a variety of encephalopathic presentations) has been confirmed worldwide The original paper additionally reported a sensitivity of 73% for NMO-IgG.22 Subsequent studies demonstrated excellent specificity, but low sensitivity (58%) for this methodology.25 ,26 Distinguishing NMO from MS clinically in the early stages of the disease is critical, since the treatments and prognosis of these disorders differ,27 and there is some evidence suggesting that MS treatments (including interferon-β) exacerbate NMO.27 ,28 Therefore, optimising NMO-IgG assay sensitivity has been the focus of recent studies.

Immunofluorescence pattern of bound neuromyelitis optica (NMO)-IgG in mouse brain and kidney tissues. (A) Linearly stained pia (P) and subpia divide a section of mouse midbrain (M) and cerebellum. Capillaries in the white and grey matter of the midbrain and cerebellum are stained, and are prominent in the central cerebellar white matter and in the granular and molecular (ML) layers. The subpial region is stained in a mesh-like pattern. (B) In the kidney, the distal collecting tubules in the renal medulla are stained. Reproduced with permission from the American Medical Association 2010.21
Early reports of AQP4-antibody assay methodologies include indirect immunofluorescence with mouse brain slices, immunoprecipitation (reported sensitivities of 33–76%),25 ,26 AQP4-transfected cell-based assays (reported sensitivity of up to 80%29 and, more recently, ELISA and flow cytometry. A recent international collaborative study compared six assay methods side-by-side.30 Assays based on binding of IgG to HEK293 cells transfected with AQP4 proved to be most sensitive: quantitative flow cytometry (77%), cell binding assays with fixed cells (73%). An ELISA assay was less sensitive (60%). GFP-AQP4 fluorescence immunoprecipitation assay and tissue-based immunofluorescence assays were least sensitive (53% and 48%, respectively). Serum testing is generally more sensitive than CSF testing; CSF testing should be reserved for patients where a high index of suspicion for an NMOSD remains despite negative serological evaluation. Although pathogenetically interesting, NMO-IgM seems to lack clinical sensitivity and specificity.31 Glial fibrillary acidic protein (GFAP) levels measured in CSF of patients in the acute phase of NMO have been observed to be markedly elevated as compared with patients with MS, indicating massive astrocytolysis in NMO,32 and might serve as a diagnostic marker of NMO in NMO-IgG seronegative patients.
The high frequency of clinical and serological autoimmune accompaniments in NMOSD further distinguish these disorders from MS. Coexisting autoimmune diseases and autoantibodies are frequent in patients with NMO, both non-organ-specific (systemic lupus erythematosus, Sjogren syndrome)33 and organ-specific (eg, thyroid disease, type 1 diabetes, celiac disease and myasthenia gravis (MG)).34–36 Antinuclear antibodies (double-stranded DNA, extractable nuclear antigen) are the most common non-organ-specific autoantibody accompaniments.33 In one study, autoantibody markers of Sjogren's syndrome or systemic lupus erythematosus were found in 47% of patients with NMOSD. Of note, however, NMO-IgG was not found in patients with Sjogren's syndrome, or systemic lupus erythematosus, in the absence of clinical features of NMO. Those cases, previously considered complications of the collagen vascular disorders, may represent, instead, the coexistence of NMO. Other organ-specific autoantibodies include thyroid peroxidase and thyroglobulin. Celiac disease-related antibodies (including deamidated gliadin and tissue transglutaminase) and antiphospholipid antibodies have also been described in some cases with NMOSD.37 ,38
Characteristic profiles of neural autoantibodies other than AQP4-antibody, both organ-specific and non-organ-specific, are detectable in the majority of patients with NMO and NMOSD, but are very infrequent in patients with MS. For instance, in one study, muscle AChR antibody was detected in 11% of NMO patients, but in no MS patients or healthy subjects.34 Clinical and electrophysiological findings consistent with MG were documented in 2% of NMO patients. Among these 4 patients, 3 had MG 11–24 years prior to onset of NMO (1 had undergone thymectomy), 2 were NMO-IgG seronegative, and 1 had thymic carcinoma. While most reported patients with both MG and NMO have a typical, severe clinical NMO course 34–36 occasional patients with MG with aquaporin-4 antibodies have been reported to have a mild NMO phenotype with subtle abnormalities on examination or paraclinical testing only.39
Pathogenesis and pathology
AQPs are tetrameric water channels composed of 30 kDa monomers, each made up of six membrane-spanning helical domains and two short helical segments. In the CNS, AQP4 regulates water, glutamate and potassium transport. Mice lacking AQP4 are less prone to cytotoxic oedema but more prone to vasogenic oedema.40 The AQP4 protein is expressed as two major isoforms: a long isoform, M1, and a shorter isoform, M23. AQP4 can form supramolecular complexes in membranes called orthogonal arrays of particles (OAPs).41 Intertetrameric N-terminus M23–M23 interactions permit large OAP formation. M1 in isolation forms few or no OAPs, because their N-termini interactions are blocked by residues upstream of Met-23.
AQP4, is highly expressed in astrocytic end-feet in the blood-brain barrier, nodes of Ranvier and neuronal synapses. In vitro studies have demonstrated that binding of NMO-IgG to AQP4 initiates multiple pathogenic mechanisms. AQP4 downregulation occurs through antigenic modulation (binding of bivalent IgG to adjacent AQP4 molecules), followed by internalisation of antigen from the plasma membrane, and targeting of antigen to lysosomal pathways.42 ,43 This occurs in parallel with downregulation of the glutamate transporter excitatory amino acid transporter 2 (EAAT-2, which is likely physically linked to AQP4) resulting in disruption of both water and glutamate homeostasis.42 Also, binding of NMO-IgG (mainly IgG1 subclass) to AQP4-expressing cells in the presence of complement, activates the classical complement cascade, leading to membrane lesioning,44 Addition of NMO patient serum to this system promotes migration of granulocytic leukocytes across the endothelial layer, natural killer cell migration and increased permeability of the endothelial barrier to plasma proteins. The binding of NMO-IgG to the ectodomain of astrocytic AQP4 has isoform-specific outcomes: M1 is completely internalised, but M23 resists internalisation and is aggregated into OAPs, that activate complement more effectively than M1 when bound by NMO-IgG. Also, NMO-IgG binding to either isoform has been shown to impair water flux directly, independent of AQP4 down-regulation.45
An interleukin 6 (IL-6)-dependent B cell subpopulation of plasmablasts may be involved in the pathogenesis of NMO. It has been shown that the plasmablast population is expanded during NMO relapse, and that IL-6 enhances their survival and their AQP4 antibody secretion, whereas the blockade of IL-6 receptor reduces the survival of plasmablasts.46 AQP4-specific T cells may also have a role in driving the AQP4-specific humoral immune response.
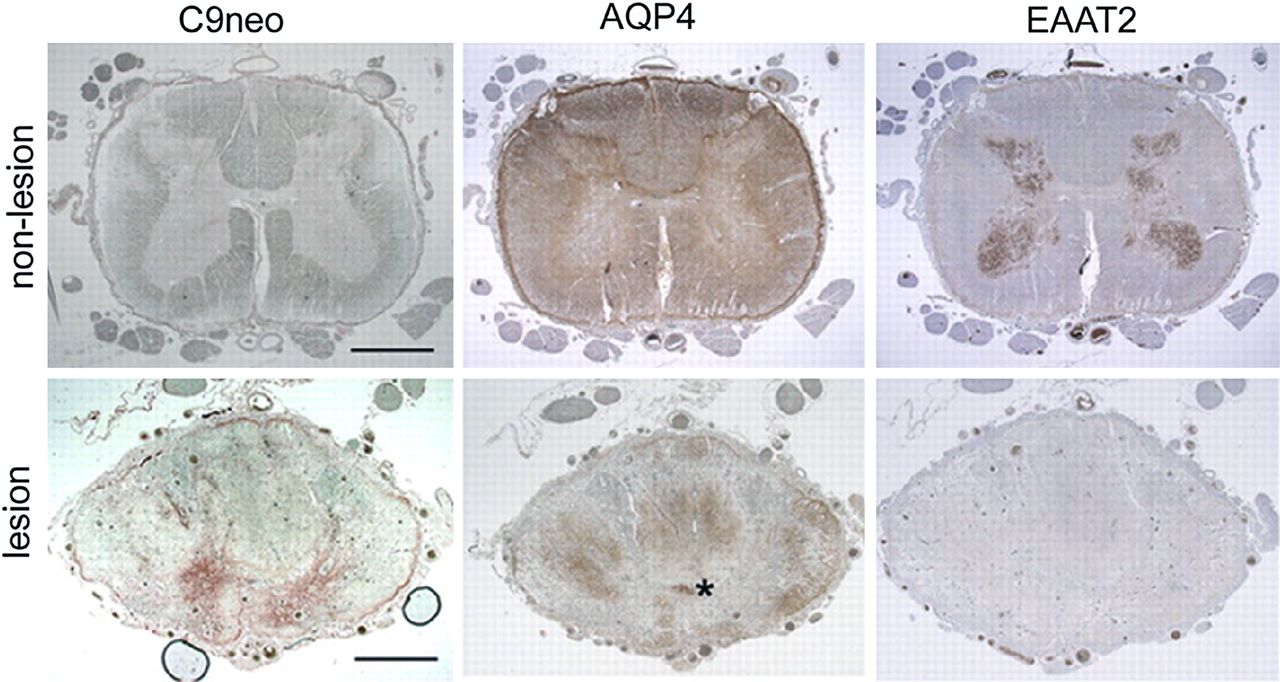
Neuropathological findings in autopsied and biopsied specimens from patients with NMO (distinct from the neuropathology of MS) have paralleled these findings of in vitro disease models (figure 3). Demyelination and necrosis are seen in both grey and WM, and blood vessels are thickened and have a pink, glassy appearance (hyalinisation).47 In active lesions, oedema and leukocytic infiltrates are prominent, with both polymorphs (eosinophils and neutrophils) and mononuclear cells (macrophages, lymphocytes and plasma cells) seen. Immune complexes (IgG, IgM and complement components) are observed around blood vessels. There is extensive loss of AQP4, GFAP48 ,49 and EAAT2,42 which appears to precede demyelination (figure 4). Impaired water flux leads to oedema, evident by the presence of vacuolation in adjacent myelin.45 Some reactive astrocytes have persistent foci of surface AQP4, consistent with isoform-specific variability in AQP4 modulation.45 The neuropathological findings in MS are in contrast with the findings in NMO; complement is deposited within macrophages or along myelin sheaths, and AQP4 loss occurs only in end-stage lesions that have undergone widespread destruction.48

Pathological and immunopathological findings in neuromyelitis optica (NMO). (A) Extensive demyelination of the grey matter and white matter at the level of the thoracic cord (Luxol fast blue-periodic acid Schiff (LFB-PAS) stain for myelin; 10×). (B) Extensive axonal injury, necrosis and associated cavitation (Bielschowsky silver impregnation; 10×). (C) and (D) The inflammatory infiltrate contains perivascular and parenchymal eosinophils and granulocytes (100×). (E) Prominent vasculocentric complement activation, in a characteristic rosette and rim pattern surrounding thickened blood vessels (immunocytochemistry for C9neo antigen (red); 200×). (F) Higher magnification (1000×) of rosette pattern of immunoglobulin deposition (immunocytochemistry for IgG). (G) Rosette pattern of C9neo antigen in a similar distribution around the same vessels as in panel F. All images were from a spinal cord lesion from a 52-year-old woman who died from active NMO associated with a longitudinally extensive spinal cord lesion extending from C3 to T8. Reproduced with permission from Elsevier 2007; Wingerchuk et al, The Spectrum of Neuromyelitis optica. Lancet Neurol 2007;6:805–15.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Spinal cord tissue from a single neuromyelitis optica-IgG-seropositive patient. Three sections of non-lesioned lumbar region (top) serve as staining control for lesioned cord (bottom). The lack of complement deposition (C9neo, brick red in lesioned cord, bottom) and high expression of AQP4 in both white and grey matter are typical of normal cord tissue; EAAT2 is highly enriched in grey matter. Prominent deposition of C9neo in grey matter of lesioned thoracic cord (bottom, same patient) corresponds to focal regions of AQP4 and EAAT2 loss in adjacent sections. Aquaporin-4 is partially retained in the white matter. Asterisk, central canal. Bar=200 μm. Reproduced with permission from Rockefeller Press, 2011.67
Some aspects of the NMO pathology have been observed in animal models, where purified IgG from AQP4-antibody-positive NMO patients was injected intravenously or intraperitoneally into rats where the blood-brain barrier was already permeabilised by the inflammatory consequences of experimental autoimmune encephalomyelitis,50–52 or where patient NMO-IgG and human complement were injected directly into the brains of mice.53 An active immunisation animal model recapitulating relapsing NMO (akin to the animal models of MG) has not been developed to date.
Treatments
There are four aspects of treatment of NMO: acute treatment of relapses, prevention of relapses, symptom management and rehabilitation.
Treatment of acute exacerbation
Once the diagnosis of NMO/NMOSD is established, acute management of a subsequent relapse is of prime importance, as disability accrues with attacks. Many patients have permanent and severe disability after the very first episode. Management of relapses is with early corticosteroid treatment, typically 1g of intravenous methylprednisolone for 5 days followed by oral prednisone (1 mg per kg body weight) for a month, and then a gradual tapering off over a 6–12 month period. Relapses that do not respond to intravenous steroids could benefit from plasma exchanges (PLEX); typically 5–7 exchanges over a 2-week period.54 ,55 The threshold to initiate PLEX should be low. The authors usually initiate PLEX in the 2nd week (after high-dose steroids) if no recovery is seen, and if deficits are severe. Since one cannot make the diagnosis of NMOSD until the AQP4 antibodies return as positive, it could be argued that the first episode of any ‘idiopathic’ LETM should be managed this way until the AQP4 antibody serological status is known. If positive, confirming the diagnosis of NMOSD, corticosteroids should be continued and additional immunosuppression considered. If negative, and no other predictor for a relapsing aetiology is found, steroids may be tapered off sooner. There is no report concerning therapeutic efficacy of intravenous immunoglobulin (IVIG) for acute exacerbation of NMO.
Prevention of relapses (table)
Considering the antibody-mediated mechanisms, treating NMO with immunosuppressant medications seems logical. However, little high-quality evidence exists for the role of the various immunosuppressant therapies. table 1 summarises the main studies of the most commonly used drugs. The rarity of the disease, severity of the relapses, early morbidity and mortality in untreated NMO, have made controlled trials difficult and placebo-controlled trials unethical. But considering the rapid accumulation of attack-related disability of untreated NMO, early initiation of treatment (in addition to corticosteroids) is now considered standard practice. Anecdotally, most immunosuppressants seem to work to varying degrees, and cost, convention, convenience, availability, side effects and familiarity of the treating physician to the drug are the guiding factors in its choice at present. There are no randomised controlled studies (RCTs) that guide therapy, and evidence is limited to case series.
Summary of common immunosuppressive medications used in NMO
The general principle of management is to quickly achieve and maintain remission with corticosteroids, choose an immunosuppressant, establish it, and then start a gradual withdrawal of corticosteroids aiming to minimise its side effects. Since the biological effects of many corticosteroid-sparing agents take months to have an effect, corticosteroids may be needed in many patients at doses 0.5–1 mg/kg for up to 3 months after an attack, and then slowly tapered off over a further 6–12 months. Some patients need a low maintenance dose of steroids to maintain remission, and this is an acceptable long-term strategy as long as there are no significant side effects. As in other immunological disorders, a detailed discussion on the potential benefits and hazards (hematological, hepatic, cardiac, renal and gastrointestinal effects, infections, malignancies and effects on reproductive function) is critical before embarking on lifelong treatment. Adequate laboratory monitoring arrangements have to be in place.
By contrast to immunosuppressants, such as azathioprine (AZA), several conventional MS drugs seem to worsen NMO. Interferon-β is now contraindicated in NMO. Many reports confirm not only its inefficacy, but the tendency to cause severe relapses; antibody levels also frequently rise in those patients.28 ,56 ,57 This may be mediated by an enhanced Th17 response.58 ,59 Both Natalizumab (Tysabri) 60 ,61 and Fingolimod (Gilenya) 62 have been reported to fulminantly exacerbate NMO. It might even be worth testing for AQP4-antibody in presumed relapsing-remitting MS that develop serious relapses following initiation of the above mentioned drugs.
Oral immunosuppressive drugs
Corticosteroids
Prednisone used alone could be effective, and is very frequently used in Japan.63 However, concerns regarding side effects prevent its continuous use as a single agent, at least in Europe and the Americas.
Azathioprine
AZA has been, for long, the principal drug in relapse prevention. Its popularity was based initially only on a short case series, but recent larger retrospective case series vindicate its use.64–66 As up to 10% of the population may lack thiopurine methyl transferase, an enzyme needed to metabolise AZA, checking its levels before treatment is good practice. Alternatively, it can be begun at a low dose and slowly built up to the usual dose of 2.5–3 mg/kg/day. Monitoring the mean corpuscular volume (MCV) for a rise of at least five points from baseline is likely a reliable way of ensuring maximisation of efficacy. Costanzi and colleagues recently demonstrated that a rise in MCV was correlated with a reduction in annualised relapse rate in patients with NMO treated with AZA.66 However, many patients relapse when attempting a complete tapering off corticosteroids, and it is reasonable to maintain such patients on a combination of AZA (or an equal) with lowest dosage of steroids (typically, alternate day steroids about 10 mg–20 mg) if side effects of corticosteroid are not a concern.
Mycophenolate mofetil
Mycophenolate mofetil is an immunosuppressive drug that blocks proliferation and clonal expansion of T and B lymphocytes. It is routinely used in organ transplantation, and is being increasingly used in a variety of other autoimmune conditions. In a 24-patient (seven treatment-naïve) retrospective series at median dose of 2000 mg/day, relapse rates were improved disability improved or stabilised in most patients.67 The convenience of oral usage, tolerability and safety are an advantage.
Intravenous drugs
Rituximab
Rituximab (RTX) as an anti-CD20 monoclonal antibody has gained popularity in the treatment of many immunological diseases. Several case series support its benefit in NMO refractory to other drugs, and repeated use has maintained efficacy.68–72 The dosing regimens of RTX are based on regimens used in rheumatoid arthritis (1 g intravenous on day 1 and day 14 repeated 6-monthly) or hematological malignancies (375 mg/m2/week for 4 weeks repeated 6-monthly). The rheumatoid arthritis regimen is perhaps easier to use, and was used in the relapsing MS trials. Monitoring of CD 19, CD27, AQP4 antibodies and BAFF (B cell activating factor) can guide treatment, though no consistent correlation is seen. An alternative to empiric 6-monthly retreatment may include on a ‘when needed’ basis, based on a panel of combined peripheral blood markers measures. Kim et al found CD27 (+) memory B cells particularly useful and retreating when they are more than 0.05% of peripheral blood mononuclear cells with 375/m2 of RTX-maintained remission.72 While costs can be prohibitive, and concerns of serious infections remain, (including progressive multifocal leukoencephalopathy) neurologists worldwide, particularly MS neurologists, with their familiarity with other monoclonal antibodies are increasingly comfortable with its use.
Mitoxantrone
Mitoxantrone is a potent immunosuppressant, has a role in refractory cases, and two case series support this.73 ,74 The major concern is that of cardiac dysfunction and therapy-related acute leukaemia which can occur in 0.2–0.8% patients, and usually at doses >60 mg/m2. The total dose is limited to 100–120 mg/m2. Subsequent immunosuppression with AZA, RTX and mycophenolate has been continued without complications.
Other drugs
IVIG, methotrexate,75 cyclophosphamide, cyclosporine, tacrolimus, glatiramer acetate76 ,77 or PLEX at regular intervals78 are other options.
Which drug to use first and how to escalate therapy?
It is hard to provide an algorithm of treatment, particularly because most immunosuppressants have a beneficial role in the majority of patients. The major question is: do all patients need drugs like RTX at the onset of the disease? Is it not reasonable to try cheaper conventional drugs like AZA with a known safety profile initially? An RCT comparing standard versus best therapy may help resolve some of these uncertainties. But for now, with rising healthcare costs, we still think AZA, with or without steroids, a good first choice. If AZA, when used as first-line agent, is not tolerated due to side effects, we use mycophenolate mofetil or methotrexate. If relapses occur on adequate first-line agents, the ‘second-line’ agents may be RTX, mitoxantrone or cyclophosphamide.
Future directions of immunosuppressive therapy
Combination therapies (eg, RTX+methotrexate, as in rheumatoid arthritis) and induction followed by maintenance regimens (eg, mitoxantrone followed by AZA/mycophenolate) can be considered. Stem cell trials are still in their infancy; three are in progress, two with haematopoietic cells and one using mesenchymal umbilical cord cells (http://www.clinicaltrials.gov/). One reported case showed no benefit.79 Humanised anti-CD20 monoclonal antibodies, ofatumumab and ocrelizumab, may avoid generation of antichimeric antibodies, and may have additional modes of action. Trial results on eculizumab (a humanised monoclonal antibody against complement fraction C5) are awaited (clinicalTrials.gov). The explosion in our understanding of the immunological mechanisms in NMO have exposed many other potential targets including glutamate receptors, Th17 blockade, BAFFs, and even specific protective monoclonal antibodies that bind AQP4 (Aquaporumab), to name a few.80
Symptom management and rehabilitation
Pain, stiffness, fatigue, bladder and bowel symptoms, have to be managed with appropriate medications. Of particular importance is tonic spasms: sudden painful spasm of upper, and /or lower limbs, that almost appears dystonic, lasting usually less than a minute. Carbamazepine around 100–400 mg/day in divided doses, often leads to rapid and gratifying relief.
Rehabilitation is an important aspect of the disease. Disabilities accrued often after the very first episodes are often irreversible. Traditionally, most NMO patients with a severe myelitis go through rehabilitation programme and then return to the community. Their follow-up then remains in neurology/MS clinics, which is perhaps not ideal. A special multidisciplinary team in a dedicated NMO clinic may be better to follow-up on these patients. Such clinics (like the UK NMO Service) should ideally have neurologist, specialist nurse, physiotherapist, occupational therapist, dietician, psychologist, continence nurses, orthoptist/ophthalmologist. Spinal cord regenerative strategies offer hope to those with permanent deficits.
Treating NMOSD
The treatment of NMOSD (assuming they follow the clinical course of NMO) should logically be similar to that of NMO. This is based on two principles: first, that there is no ‘benign’ NMO.81 Second, if AQP4-antibodies are present, then relapses are invariable at some point. Both these concepts have some evidence (retrospective series) to support them. The opposite has not been reliably confirmed, though there must be occasional cases that are in ‘remission’ for many years. So, as the vast majority of patients are expected to relapse with the potential for permanent disability, we discuss the pros and cons of treatment and suggest immunosuppressive therapy. If patients opt not to start treatment, clear plans for an early and aggressive management of a relapse (steroids, PLEX) are put in place, and patients educated to seek help at the earliest.
Untreated or inadequately treated typical NMO has a poor prognosis, at least from retrospective studies. More than half the patients developed severe visual loss in at least one eye, or had inability to ambulate without assistance within 5 years of disease onset with 5-year mortality rates in relapsing patients of 32%.16 However, more recent data, including that in the new era of NMO IgG testing and early treatment, have been encouraging. In a recent British–Japanese study, only 10/106 (9%) had died at last follow-up; the median time to death was 100 months, and NMO was the cause of death in only 7/10.82 In a German cohort, only 9/175 (6%) patients had died at the end of the observation period; only five of these patients died from NMO-related causes.8
Conclusion
Our understanding of NMO has rapidly changed in the last decade, and the next few years should provide further exciting insights. Translating this new knowledge into clinical therapeutic trials is the next step. The bigger challenge is to use ‘codes that we have unlocked’ in NMO to understand other demyelinating disorders, particularly MS.
References
Footnotes
-
AJ, AM and IN contributed equally.
-
Contributors JDS authored the introduction, IN and DKS authored the sections on epidemiology, clinical features and MRI findings. AM authored the sections on NMO-IgG (AQP4-antibody) and other autoantibody findings, and pathogenesis and pathology. AJ and LE authored the sections on treatment. All authors contributed to acquisition of references and data, analysis and interpretation of data, drafting and revising the manuscript and approved the final version of the manuscript.
-
Funding (1) This study was supported by grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology (20390241, 21790828, 22229008), and the Ministry of Health, Labour and Welfare of Japan. (2) The NMO service in the UK is funded by the National Health Service, through the National Specialised Services Commissioning Group.
-
Competing interests Dr Fujihara has received honoraria for speaking or consulting from Asahi Kasei Medical, a manufacturer of devices for apheresis.
-
Provenance and peer review Commissioned; externally peer reviewed.