Article Text

Abstract
Background and purpose Cerebral amyloid angiopathy (CAA) is common in the ageing brain and is associated with dementia and lobar intracerebral haemorrhage. We systematically reviewed genetic associations with CAA to better understand its pathogenesis.
Methods We comprehensively sought and critically appraised published studies of associations between any genetic polymorphism and histopathologically confirmed CAA. We assessed the effects of genotype by calculating study specific and pooled odds ratios (ORs) in meta-analyses, and assessed small study bias.
Results 58 studies (6855 participants) investigated apolipoprotein E (APOE) genotype and sporadic CAA. Meta-analysis of 24 (3520 participants) of these showed an association of APOE ɛ4 with CAA (ɛ4 present vs absent, pooled OR 2.7, 95% CI 2.3 to 3.1, p<0.00001), which was dose dependent, robust to potential small study biases and occurred irrespective of dementia status. There was no significant association between APOE ɛ2 and CAA. Among 24 studies (4703 participants) of other genetic polymorphisms, there was preliminary evidence of an association with CAA of polymorphisms in the transforming growth factor β1 gene (two studies, 449 participants), translocase of outer mitochondrial membrane 40 gene (one study, 723 participants) and the complement component receptor 1 gene (one study, 544 participants). There were insufficient data to draw conclusions from 24 studies (∼200 participants) of APOE and hereditary CAA or familial Alzheimer's disease.
Conclusions There is convincing evidence for a dose dependent association between APOE ɛ4 and sporadic CAA. Further work is needed to better understand the mechanism of this association and to further investigate other genetic associations with CAA.
- Amyloid
- Cerebrovascular Disease
- Neurogenetics
Statistics from Altmetric.com
Introduction
Cerebral amyloid angiopathy (CAA) is characterised by peptide deposition, mainly amyloid β, in cortical and leptomeningeal arteries. CAA is associated with increasing age, dementia, lobar intracerebral haemorrhage (ICH), lobar brain microbleeds, leukoaraiosis, small cortical infarcts and superficial siderosis.1–4 ICH is usually attributed to CAA when pathological examination reveals extensive CAA deposition and related vasculopathic changes.5
Identifying genetic polymorphisms associated with histopathologically confirmed CAA should increase understanding of the mechanisms leading to CAA and associated diseases. Polymorphisms in the apolipoprotein E gene (APOE)6 are associated with many conditions in which CAA may be involved, including subarachnoid haemorrhage, ICH, lobar brain microbleeds and Alzheimer's disease (AD).4 ,7–12 In vitro studies have shown that APOE influences Aβ conformation, fibril formation and toxicity13 ,14 while in vivo mouse studies have confirmed a critical role for APOE in Aβ deposition, toxicity and possibly clearance.15 ,16 The currently favoured view is that APOE ɛ4 enhances deposition of amyloid β in cerebral blood vessel walls while ɛ2 promotes haemorrhage from amyloid laden blood vessels by increasing specific CAA related vasculopathic changes.17–19
A clear association between APOE and histopathologically confirmed CAA would explain many of the observed associations between APOE and clinical outcomes. There is robust large scale evidence for an association of APOE with ICH attributed to CAA on the basis of clinical criteria. However, studies suggesting an association of APOE with histopathologically confirmed CAA have been limited by various methodological shortcomings, including small size. Furthermore, the possibly contrasting effects of the different APOE alleles on CAA have remained unclear. No systematic evaluation has been performed of the association of APOE or of other polymorphisms with histopathologically confirmed CAA. We therefore aimed to assess the evidence for associations between genetic polymorphisms in any gene and histopathologically confirmed CAA by carrying out a systematic review, incorporating a comprehensive search strategy, a thorough assessment of study quality, a series of meta-analyses and an evaluation of the robustness of any positive findings to small study and other methodological biases.
Methods
Study identification and inclusion/exclusion criteria
We sought all studies of adult humans published in any language in which participants had been genotyped for any genetic polymorphism and had CAA assessed pathologically (using autopsy or biopsy), regardless of whether any association was reported on.
We searched OVID Medline (1950 to March 2012) and Embase (1980 to March 2012) using a combination of search terms for APOE, genes and CAA (see online supplementary appendix S1). We also checked the bibliographies of all relevant studies and reviews identified, and searched Google Scholar for studies citing relevant studies.
We wished to study genetic associations across the range of CAA severity and so included studies of genetic associations with: the presence versus absence of CAA pathology; more versus less severe CAA; or average CAA score. We excluded studies that assessed genetic associations with CAA associated ICH (CAAH) versus CAA free controls because these would not be able to distinguish a genetic association with CAA from an association with ICH. However, we included studies that had recruited participants with CAAH as an unselected part of the spectrum of CAA severity.5 Two authors independently selected eligible studies, resolving disagreements by discussion.
We classified studies into three partly overlapping groups, according to the association investigated: APOE and sporadic CAA; APOE in any type of hereditary CAA (HCAA) or familial Alzheimer's disease (FAD); any other (non-APOE) genetic polymorphism and sporadic CAA. When two or more studies included overlapping sets of participants, we included only the study providing most data about the association among the largest number of participants. To avoid undue effort on relatively small studies, we further excluded studies with <35 participants from the APOE and sporadic CAA group thus excluding <3% of the total participants in eligible studies in this group.
Data extraction
APOE and sporadic CAA studies
For each study included, we extracted information on: first author; publication year; country in which the study was conducted; and participant source and characteristics. We also assessed the quality of reporting of genotyping based on the STREGA (Strengthening the Reporting of Genetic Association Studies) and MOOSE (Meta-analysis of Observational Studies in Epidemiology) recommendations,20 ,21 and the quality and characteristics of CAA pathology assessment (see online supplementary methods).
Where possible, we extracted data on the numbers of participants with ɛ4 or ɛ2 allele containing (ɛ4+ or ɛ2+) genotypes and their CAA status, in dichotomous (present or absent) or continuous (mean (and SD) severity score) format. In studies without such data available, we recorded any qualitative statement about the association between APOE and CAA. At least two authors extracted these data, resolving disagreements by discussion.
Studies of APOE in HCAA/FAD and of other genetic polymorphisms and sporadic CAA
For these smaller groups of studies, we extracted information on: first author; publication year; number of participants; polymorphism(s) studied; and any data about the association between the polymorphism studied and CAA.
Statistical analyses (APOE and sporadic CAA)
We used Cochrane RevMan (V.5) software. For studies presenting data in a dichotomous format, we calculated an unadjusted OR for the presence versus the absence of CAA among those with ɛ4+ versus ɛ4− and ɛ2+ versus ɛ2− genotypes. For studies presenting continuous data, we calculated standardised mean differences (SMD) in CAA scores. Then, to analyse the data from dichotomous and continuous studies together, we used the method described by Chinn to convert SMDs to ORs.22 We also compared ɛ4 homozygous and heterozygous genotypes. We calculated pooled ORs using the generic inverse variance fixed effects method, and repeated all analyses using a random effects model. We assessed heterogeneity with I2 and χ2 statistics. We considered p<0.05 to imply statistical significance.
We assigned each included study a quality score, based on study size, participant recruitment method (prospective vs other), blinding, quality of genotyping and pathology assessment, and data format (considering continuous data superior due to potential bias in selecting cutoffs for dichotomous data). We conducted prespecified subgroup analyses based on: dementia status; ethnicity; study quality score; and each quality criterion separately. For any statistically significant result, we assessed the potential effect of unpublished negative studies or studies not reporting the data required for meta-analysis (ie, publication and reporting biases), using a modified ‘failsafe N’ method: we determined the size of a notional study with a null result (OR=1) required to bring any significant result (p<0.05) to a just non-significant level (p=0.05), assuming the overall prevalence of CAA and distribution of genotypes to be the average of these for the studies included.23 We used the size of this notional study as a guide to whether there might plausibly be enough participants in unpublished, unreported or otherwise unretrieved null studies to make an apparently significant result non-significant if data from such studies were available for inclusion in our meta-analyses. We also inspected funnel plots.
Results
Of 1754 publications identified for screening, 136 were relevant (figure 1).
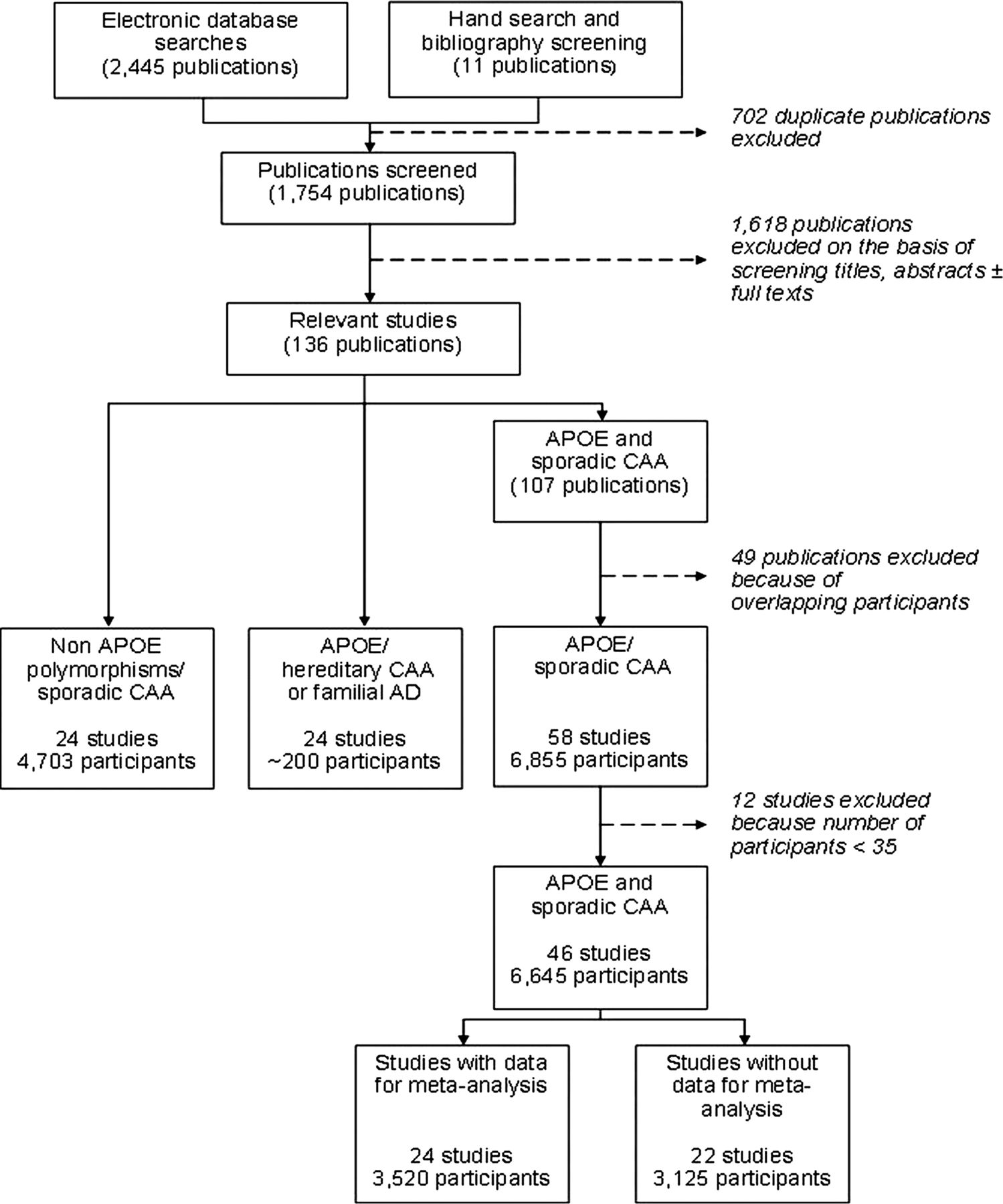
Selection of included studies. AD, Alzheimer's disease; APOE, apolipoprotein E; CAA, cerebral amyloid angiopathy.
Studies of APOE ɛ2/ɛ3/ɛ4 polymorphism and sporadic CAA
From 107 relevant publications we excluded 61 (49 because they included participants overlapping with included studies and 12 because of very small size (< 35 participants)),s1–s61 leaving 46 studies, including 6645 participants overall (figure 1).19 ,24 s62–s105 (See online supplementary tables S1b, S1c and results.)
Study characteristics and quality
All studies used autopsy brains from a brain bank, clinical autopsies or a population based prospective study. The median number of participants per study who were both genotyped and assessed pathologically was 100. Mean age was 70–85 years in most studies. About half of all participants were male. Nineteen studies (2660 participants) were conducted in white populations in Europe, 21 (3225 participants) in white subjects in the USA, five (714 subjects) in Asian populations (three in Japan, two in Japanese–Americans in the USA) and for one study this information was unavailable. About 30% of participants had clinical dementia (mainly AD), about 10% were known not to be demented and dementia status was not specified for about 60% (see online supplementary table S1a).
Genotyping quality was generally limited when assessed against current reporting standards,20 ,21 and methods for pathological assessment were very variable (see online supplementary tables S1b, S1bc and results). There was substantial variation in overall study quality. For the studies in the meta-analysis, we calculated quality scores based on our pre-set criteria. No study fulfilled all of the criteria but larger studies tended to have higher quality scores (table 1).
Study quality scores
Association between APOE ɛ4 and CAA
Twenty-four studies out of 46 (3520 of 6645 participants) provided sufficient quantitative data for meta-analysis (14 dichotomous and 10 continuous format) (figure 2). Twenty-two studies could not be included in our meta-analysis: six (443 participants) of these made a qualitative statement while 16 (2682 participants) provided no data about the association.

Meta-analysis of association of apolipoprotein E ɛ4+ versus apolipoprotein ɛ4− genotypes with cerebral amyloid angiopathy (CAA) by participants’ dementia status. The squares represent study specific ORs, with their size proportional to their statistical weight by the generic inverse variance method. Horizontal lines represent 95% CIs. Diamonds represent pooled ORs and their width represents 95% CI.
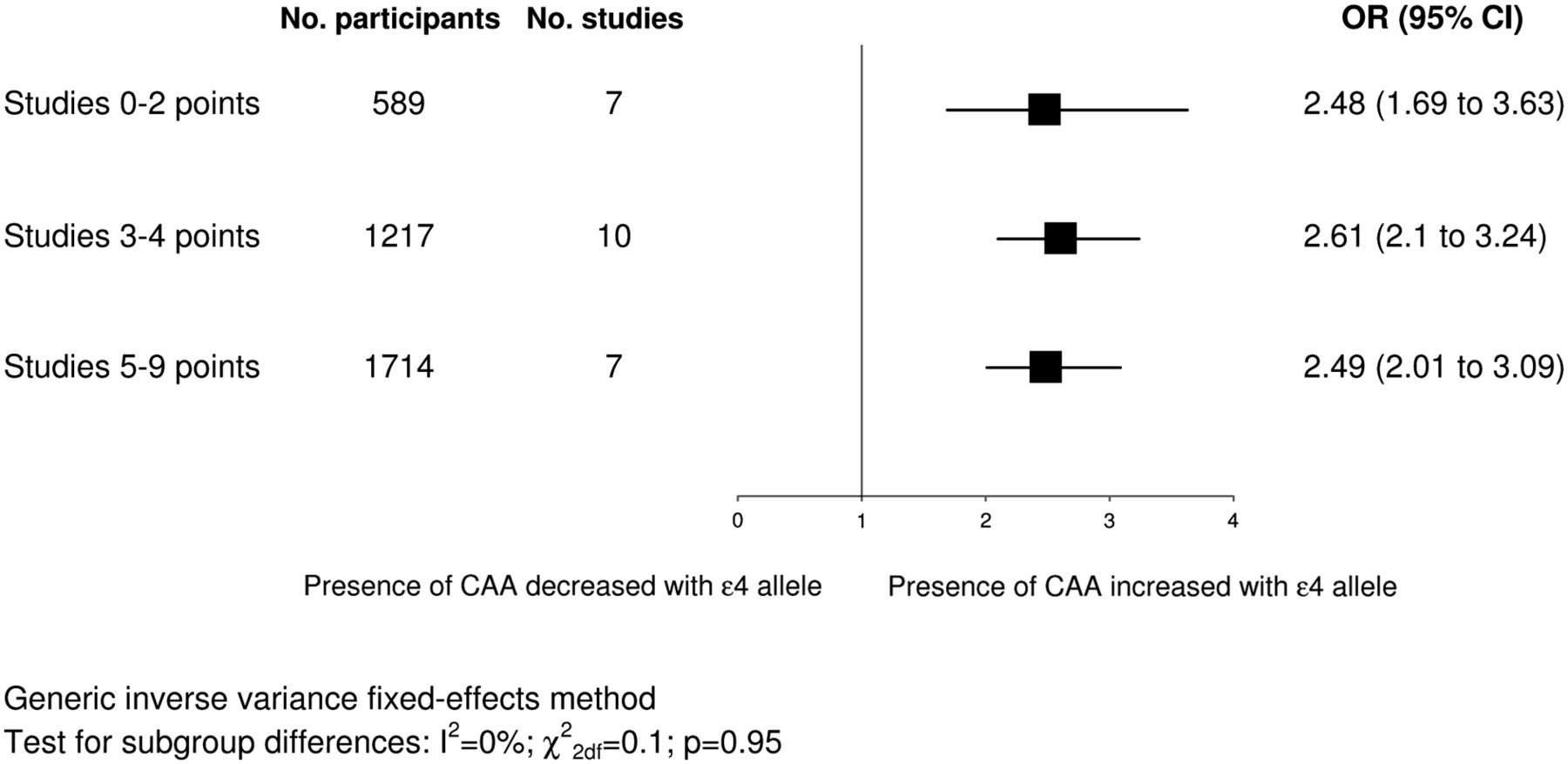
Figure 2 shows study specific and pooled ORs for ɛ4+ versus ɛ4− genotypes. The pooled OR showed a significantly increased odds of having CAA for ɛ4+ genotypes (OR 2.67, 95% CI 2.31 to 3.08) with significant heterogeneity between the results of the studies (I2=69%; χ228df=89; p<0.00001). When we meta-analysed studies providing continuous and dichotomous data separately, pooled results were in the same direction as the overall pooled OR and were separately significant (continuous studies: SMD 0.48, 95% CI 0.38 to 0.59, p<0.00001; dichotomous studies: OR 4.15, 95% CI 3.23 to 5.34, p<0.00001). Subgroup analysis based on dementia status showed no significant subgroup differences (I2=0%; χ22df=1.91; p=0.38), with similar results when we limited the dementia subgroup to neuropathologically verified AD cases only. We detected no significant differences in subgroup analyses based on ethnicity (data not shown). The association did not vary significantly by study quality score (figure 3). Nor did we detect significant subgroup differences based on the individual quality criteria of study size, blinding or quality of genotyping. The association was slightly larger in studies with prospective recruitment of participants (OR 3.66, 95% CI 2.87 to 4.68 vs OR 2.56, 95% CI 2.15 to 3.05; I2=82%; χ21df=5.5; pdiff=0.02), in studies presenting data in a dichotomous versus a continuous format (OR 3.64, 95% CI 2.91 to 4.55 vs OR 2.48, 95% CI 2.06 to 2.98; I2=85%; χ21df=6.8; pdiff=0.009) and in studies with higher quality of pathology assessment (2 points: OR 3.07, 95% CI 2.46 to 3.83 vs 1 point: OR 2.22, 95% CI 1.84 to 2.67; I2=80%; χ21df=4.9; pdiff=0.03).

Subgroup analysis based on study quality scores. The squares represent pooled ORs and their width represents the 95% CI. CAA, cerebral amyloid angiopathy.
Failsafe N calculations showed that a null study of >137 000 participants would be required to bring the association of ɛ4+ genotypes with CAA to a just statistically non-significant level (ie, p=0.05). The funnel plot was only slightly asymmetrical. Studies providing only qualitative data reported either no significant association or a trend towards association with APOE ɛ4 (see online supplementary table S2).s62–s67
Twelve studies (1706 participants) provided quantitative data for meta-analysis of the association of APOE ɛ4 allele dose with CAA, showing a significant increase in the odds of having CAA with increasing dose of the ɛ4 allele (figure 4). Two studies (117 participants) providing a qualitative statement about the association supported this result (see online supplementary table S2).s67,s102 Failsafe N calculations showed that it would require a null study of >7000 participants to bring the stronger association with CAA of ɛ4 homozygous versus heterozygous genotypes to a just non-significant level.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Meta-analysis of effects of apolipoprotein E ɛ4 dose (ɛ4−−/ɛ4+−/ɛ4++ genotypes) on the presence versus the absence of cerebral amyloid angiopathy (CAA). Notation as for figure 3.
Association between APOE ɛ2 and CAA
Eleven studies (1640 participants) provided quantitative data for meta-analysis of APOE ɛ2+ versus ɛ2− genotypes with CAA. The pooled OR showed a non-significantly decreased odds of CAA with APOE ɛ2+ genotypes (OR 0.73, 95% CI 0.53 to 1.00, p=0.05). When we meta-analysed studies providing continuous and dichotomous data separately, pooled results were in the same direction as the overall pooled OR but remained non-significant. Two studies (213 participants) provided a qualitative statement; neither reported a significant association (see online supplementary table S2).s96,s102
Random effects meta-analyses
Results were very similar when all of the above meta-analyses were repeated using a random effects model.
Studies of APOE ɛ2/ɛ3/ɛ4 polymorphism in cases of HCAA /familial AD
Of 24 relevant studies,s60,s106–s128 only two reported on the association between the APOE genotype and severity of CAA. They found no effect of APOE ɛ4 on CAA in 16 HCHWA-D (hereditary cerebral haemorrhage with amyloidosis, Dutch type) patientss106 or in 54 participants with FAD due to a presenilin-1 mutation.s107
Studies of other genetic polymorphisms and sporadic CAA
Few other polymorphisms had been studied in more than a few hundred participants or in more than one study and there were insufficient data for formal meta-analyses.s4,s6–8,s11–13,s24,s35,s37–39,s41,s46,s71,s80–81,s97,s100,s129–133 However, there were some positive associations with CAA: a consistent trend to association with a single nucleotoide polymorphism (SNP) in the transforming growth factor β1 (TGF-β1) gene (two studies, 449 participants)s13,s24; significant associations with SNPs in the translocase of outer mitochondrial membrane 40 (TOMM40) gene (one study, 723 participants);s132 and with a SNP in the complement component receptor 1 (CR1) gene (one study, 544 participants).s133 Other studies found no overall significant associations although some reported associations in particular subgroups (table 2).
Summary of studies of non-apolipoprotein E polymorphisms and cerebral amyloid angiopathy
Discussion
To our knowledge, this is the first systematic review and meta-analysis of associations between genetic polymorphisms and pathologically proven CAA although there have been meta-analyses of genetic associations with deep and lobar ICH, ICH severity and outcome, and brain microbleeds.9–11 We found a highly significant dose-dependent association between APOE ɛ4 and pathologically proven CAA, which did not vary significantly with dementia status, ethnicity or study quality. We found no overall association between APOE ɛ2 and the presence of CAA. No modifying effects of APOE on CAA were reported among cases of HCAA or of FAD, but because most of these participants have severe CAA anyway, modulation by APOE is very difficult to detect.s109 Finally, there were too few studies and participants to draw firm conclusions about the effect of other genetic polymorphisms but there were some positive associations which merit replication in further larger studies or pooled datasets.
The prevalence of CAA in AD is over 70% but the relationship between CAA and AD is still poorly understood. Although the diagnostic criteria for dementia and the participant inclusion criteria varied between studies (with some excluding cases with severe dementia), demonstration of a similar association in those with and without clinical dementia suggests that the association of APOE ɛ4 with CAA is independent of its known association with dementia (mainly AD). This is further supported by similar results when we limited the dementia subgroup to neuropathologically verified AD cases only. Relative to other apoE isoforms, apoE4 is thought to increase aggregation or impair clearance of amyloid β, or both. While this mechanistic pathway is probably common to both CAA and AD, specific mechanisms might also occur. For example, apoE isoform specific neurotoxic effects may contribute to neurodegeneration in AD, independent of interactions with amyloid β.25
Our study benefited from thorough ascertainment and critical appraisal of pertinent studies including a large number of participants. Lack of variation in the effect of APOE ɛ4 by study size and the very large failsafe N show that this association cannot plausibly be explained by publication, reporting or any other small study bias. In addition, although study quality was generally limited when assessed against current reporting standards,20 ,21 there were—reassuringly—no significant subgroup differences by study quality score.
There were some limitations. Pathological assessment was very variable. Indeed, there is no widely accepted standardised histopathological grading system for CAA,26 and no comparative studies to determine the most accurate method for assessing CAA (although the suggested method is a combination of thioflavin S/T or Congo Red with immunohistochemistry).27 CAA assessment location varied widely, possibly influencing the rate of CAA detection as a greater burden of CAA is generally reported in the occipital or parietal lobes, albeit with a higher frequency of frontal lobe involvement reported in studies from China and Japan.26 Most included studies did not address the issue of potential confounding due to population stratification. However, such confounding is unlikely because these were not case control studies but cross sectional studies across a range of CAA severities. Finally, there was a large number of studies that we could not include in the meta-analysis because they did not report the necessary quantitative data about the association between the genetic polymorphism and CAA. However, our systematic review is strengthened by the identification and detailed characterisation of these studies. By contrast, many meta-analyses do not account for otherwise relevant studies without the necessary data within their publications and so risk undetected reporting bias as many studies report only significant findings and fail to mention non-significant ones. We have shown that our findings are robust to these missing data as our large failsafe N (>137 000) greatly exceeded the total number of participants excluded due to lack of available data (3125).
Several outstanding issues require more research. Genes other than APOE seem likely to influence CAA. Some preliminary positive findings merit replication in larger studies: the possible reported association between the TGF-β1 gene and CAA may be through an influence on Aβ clearance and deposition through activation of astrocytes and microglia; association of TOMM40 gene polymorphisms with vascular amyloid burden but not CAAH could be through interaction with APOE ɛ2 or affects on Aβ mitochondrial transport; association of a CR1 gene polymorphism with both CAA severity and CAAH may be via clearance of Aβ peptide.28 s13 s132 As the effects of APOE on ICH may vary with ethnicity, there may be ethnic variation in genetic associations with CAA but these have not been widely studied in non-white populations.29 Genetic associations may also differ by CAA location and subtype. For example, there is preliminary evidence that APOE ɛ4 may be associated with CAA type 1 (where CAA is found in cortical capillaries) and ɛ2 with CAA type 2 (where amyloid is deposited in leptomeningeal and cortical vessels with the exception of cortical capillaries).24 Finally, the suggestion of different genetic influences on amyloid deposition in the vessel wall and advanced vasculopathic changes leading to ICH requires further investigation, in particular the proposed differential effects of ɛ4 and ɛ2 alleles.
In conclusion, despite study quality issues, a large body of evidence supports an association of APOE ɛ4 with the presence and severity of histopathologically confirmed CAA, at least in white populations. Future research efforts require methodologically robust studies adhering to current reporting standards, facilitating comparisons between studies and collaborative data pooling efforts. These should focus on the differential effects of APOE ɛ2 and ɛ4, variation in genetic effects by ethnicity and CAA location, and on the potential influence of other polymorphisms on this clinically important but as yet incompletely understood phenotype.
Acknowledgments
The authors thank Lee Murphy for his assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Contributors KR: substantial contribution to the design and conceptualisation of the study, acquisition, analysis and interpretation of the data, drafting and revising the manuscript, and final approval of the version to be published. NS: contribution to the design and conceptualisation of the study, acquisition, analysis and interpretation of the data, revising the manuscript and final approval of the version to be published. NAM-G: contribution to the design and conceptualisation of the study, acquisition, analysis and interpretation of the data, revising the manuscript and final approval of the version to be published. RA-SS: contribution to the design and conceptualisation of the study, acquisition, analysis and interpretation of the data, revising the manuscript and final approval of the version to be published. CLMS: senior supervision and coordination of the study, substantial contribution to design and conceptualisation of the study, acquisition, analysis and interpretation of the data, drafting and revising the manuscript and final approval of the version to be published.
-
Funding European Federation of Neurological Societies Scientific Fellowship (KR); UK Medical Research Council research training fellowship (G0900428, NS); ConacYt-Mexico (NAM-G); UK Medical Research Council senior clinical fellowship (G1002605, RA-SS); and Scottish Funding Council (CLMS).
-
Competing interests None.
-
Ethics approval In accordance with National Research Ethics Service and Medical Research Council guidance, this study does not require ethics approval as it does not directly involve human participants. The data used in the systematic review and meta-analysis have come from studies which have previously satisfied regulatory requirements and been peer reviewed.
-
Provenance and peer review Not commissioned; externally peer reviewed.