Article Text

Abstract
The clinical approach to patients with amyotrophic lateral sclerosis (ALS) has been largely modified by the identification of novel genes, the detection of gene mutations in apparently sporadic patients, and the discovery of the strict genetic and clinical relation between ALS and frontotemporal dementia (FTD). As a consequence, clinicians are increasingly facing the dilemma on how to handle genetic counselling and testing both for ALS patients and their relatives. On the basis of existing literature on genetics of ALS and of other late-onset life-threatening disorders, we propose clinical suggestions to enable neurologists to provide optimal clinical and genetic counselling to patients and families. Genetic testing should be offered to ALS patients who have a first-degree or second-degree relative with ALS, FTD or both, and should be discussed with, but not offered to, all other ALS patients, with special emphasis on its major uncertainties. Presently, genetic testing should not be proposed to asymptomatic at-risk subjects, unless they request it or are enrolled in research programmes. Genetic counselling in ALS should take into account the uncertainties about the pathogenicity and penetrance of some genetic mutations; the possible presence of mutations of different genes in the same individual; the poor genotypic/phenotypic correlation in most ALS genes; and the phenotypic pleiotropy of some genes. Though psychological, social and ethical implications of genetic testing are still relatively unexplored in ALS, we recommend multidisciplinary counselling that addresses all relevant issues, including disclosure of tests results to family members and the risk for genetic discrimination.
- ALS
- GENETICS
Statistics from Altmetric.com
Introduction
In the last few years, the clinical approach to patients with amyotrophic lateral sclerosis (ALS) has been modified by the identification of novel genes, detected also in apparently sporadic cases, and the discovery of ALS relationship with other neurodegenerative diseases, in particular frontotemporal dementia (FTD). Furthermore, as for all other genetic predisposition tests, genetic testing in ALS may also become increasingly important at the light of the future development of novel effective therapies for specific genes.1
Patients and families, increasingly aware of the genetic component of ALS, often ask about genetic testing, even without an apparent family history. Requests are also made by at-risk subjects, namely, relatives of ALS patients or, less frequently, for prenatal diagnosis. Neurologists need help in handling the complexities underlying these requests, since often decisions are made on individual basis. The European Federation of Neurological Societies (EFNS)2 has issued guidelines indicating that DNA analysis should be limited to ‘cases with a known family history of ALS, and in sporadic ALS cases with the characteristic phenotype of the recessive D90A mutation’ and not performed in ‘cases with sporadic ALS with a typical classical ALS phenotype’. EFSN guidelines state also that ‘asymptomatic at-risk genetic testing should only be performed in first-degree adult blood relatives of patients with a known gene mutation, on a strictly voluntary basis and following accepted ethical principles.’
Criteria for the classification of familial ALS have been recently proposed.3 They are based both on the presence of first-degree or second-degree relatives with ALS and on the finding of genetic mutations. The identification of other neurodegenerative disorders, in particular FTD, in patients pedigree, is an additional criterion (see online supplementary table 1).4 Despite this classification, there is no consensus among researchers about the definition of familial cases.5
It has been advocated that the clinical genetic approach in ALS can be modelled on the example of Huntington disease (HD).6 However, while HD is a monogenic disease (albeit with modifying genes), usually with a positive family history and a rather predictable penetrance, ALS is a polygenic disorder with insufficient data on penetrance and pathogenetic nature of many mutations. Furthermore, most patients do not have a positive family history for ALS or other neurodegenerative disorders.
This document is the result of a workshop on ALS genetic testing in the clinical setting, held in Corteranzo, Italy, 20–21 September 2012, by the ITALSGEN Consortium. Participants were neurologists, geneticists, psychologists and ethicists, who focused on producing a set of clinical, rather than research, suggestions to optimise the communicative process of genetic risk to ALS patients, including discussion of medical, psychological, social and ethical implications.
Notably, reimbursement policies for genetic testing and counselling vary in countries with a different health system, also on the basis of the test required. In many countries, genetic testing in apparently sporadic cases is not reimbursed. In the USA, private insurances may deny reimbursement for genetic testing or counselling, especially in the absence of effective preventive or therapeutic measures. By contrast, in countries with socialised medicine, such as Italy, the public health system covers most instances of genetic testing and counselling, when requested by a specialist and considered clinically relevant. Yet, since the basic principles of genetic counselling, including its non-directiveness, are similar across cultures, our clinical suggestions, while born in the Italian context, are not limited to it and are intended for professionals involved in ALS care.
The disease
ALS is a neurodegenerative disorder of adult life, characterised by a progressive involvement of upper and lower motor neurons at spinal and bulbar level. The diagnosis of ALS remains clinical, supported by paraclinical exams.2 ,7 Currently, no specific biomarkers of the disease are available, the only exception being the finding of mutations of ALS-related genes and reduced SOD1 dismutation activity.7 The only defined risk factors are older age and male gender. No ‘strong’ environmental factors have been identified, possible exceptions being cigarette smoking and slim body constitution.8 While ∼90% of patients appear sporadically in the population, ∼10% of cases are familial in nature.4
ALS genes: novel discoveries and new uncertainties
The list of ALS-related genes is rapidly increasing, mainly due to the application of new technologies such as exome sequencing, but also in relation to expansion of ALS DNA banks throughout the world. About two-thirds of familial cases are caused by four genes, C9ORF72, SOD1, TARDBP and FUS.4 ,9 At least 12 other less common genes and loci are known (see online supplementary table 2); for the remaining cases, the genetic cause is still unknown. However, wide areas of uncertainty persist also with the best known gene mutations.
Frequency of ALS-related genes
The frequency of ALS-related genes is unevenly distributed in different populations. For example, SOD1 mutations account for ∼10% of ALS cases in Sweden and Finland, with the p.D90A homozygous mutation being by far the most common missense mutation,10 while they are found in less than 1% of Dutch patients.11 Similarly, C9ORF72 is the commonest gene in Caucasian populations,12 but is less frequent in patients of Chinese or Japanese ancestry.13 ,14
Are all mutations pathogenic?
In many cases, we do not have a clear evidence of the pathogenic role of some mutations. Even for SOD1, the most widely studied gene, the demonstration of pathogenic nature of some of the reported missense, nonsense and deletion mutations is poor.10 A recent paper described four familial ALS pedigrees discordant for two SOD1 mutations (p.D90A and p.E100 K), raising questions about the heterogeneity of ALS in individual pedigrees and the pathogenic nature of p.E100 K mutation.15 In the case of C9ORF72, the limitation comes from laboratory techniques. Its pathogenic mutation is a GGGGCC hexanucleotide expansion in the first intron of the gene. According to data from Southern blot analysis, the number of repeats is up to more than 1000.16 However, in most laboratories the expansion is searched with Sanger technology, not allowing to identify more than ∼80 repeats.
How high is the penetrance of gene mutations?
In general, one problem in determining penetrance is that the age-dependent likelihood of phenotypic manifestation of a genetic mutation is often drawn from a population sample of pooled data. When confronted with individual patients and their families in a clinical setting, these data are difficult to apply, as each pedigree carries a specific penetrance, which may shift across generations, due to marriage, other genetic modifiers or anticipation phenomenon.
Additionally, specific data on the penetrance of different mutations of ALS-related genes are still limited. For SOD1, some mutations have a high penetrance (eg, p.A4V, homozygous p.D90A) and others low penetrance (p.N19S, heterozygous p.D90A, p.I113T).10 Several missense and nonsense mutations have been described in single families or isolated patients, preventing the possibility of drawing firm conclusions about their penetrance. Mutations in the FUS gene seem to have quite a high penetrance, while those of TARDBP probably vary according to the involved codon. Data on C9ORF72 hexanucleotide expansion are now starting to accumulate. A penetrance rate of 50% at 60 years, and an almost full penetrance at 80 years have been estimated.12 Yet these figures should be considered with caution since no systematic assessment has been performed in large numbers of healthy controls of different ancestry. Two other areas of uncertainty are whether a premutation size does exist, and whether an anticipation phenomenon is present, similarly to what is reported for other diseases with repeat expansion.17
Is ALS an oligogenic disease?
Mutations of two ALS-related genes in the same patient are increasingly described both in familial and apparently sporadic patients,18 ,19–21 suggesting, at least in some cases, that multiple genetic factors are necessary to develop ALS or determine its phenotype.
Is there a genotypic–phenotypic correlation in ALS-related genes?
Familial and sporadic ALS cases are clinically indistinguishable, although some differences between gene-related phenotypes have been identified. For example, C9ORF72 than any other gene, is more frequently associated with comorbid FTD and psychotic-like symptoms both in the index case and in the extended family. Several other ALS-related genes may cause disorders different from ALS (see online supplementary table 2). FUS mutations are often characterised by a young age at onset (<35 years), a rapid clinical course and death occurring within 18 months.9 SOD1 mutations are extremely heterogeneous, the onset varying between the first and the eighth decade, and rate of progression going from very rapid (p.A4VV) to exceptionally slow (p.G93D, p.L144F).10 Therefore, with few exceptions, the clinical picture of a single patient cannot guide the genetic analysis, unless the gene has been already found in other family members.
Are there susceptibility genes for ALS?
In sporadic patients, several genes and loci have been proposed to be related to an increased risk, or to modify onset or progression. Only few of them have been replicated in independent studies and in different populations, the most notable being mutations of ATXN2, UNC13A, ANG, SMN1 and SMN2.22 Reasons not to offer genetic testing for these mutations in a clinical setting are: (1) none of them is necessary or sufficient to cause ALS, (2) molecular tests have low sensitivity and specificity, (3) there are no feasible preventive options at present and (4) it is difficult to correctly convey a probabilistic risk to patients and relatives with such high uncertainty.
Familial and sporadic ALS: does this dichotomy still hold?
Classically, ALS cases have been distinguished between those with a positive family history for ALS and those without other cases in the family. However, this distinction does not hold anymore. First, patients with and without genetic mutations do not show any clear-cut clinical difference, with the possible exception of those carrying the C9ORF72 expansion, who are often associated with FTD, psychotic-like symptoms and extrapyramidal features. Second, pathogenic mutations of ALS-related genes have been found, up to 10% in some series,19 in apparently sporadic ALS; a lack of family history is ascribed to several factors, namely a non-informative family history, misdiagnosis of a relative, early death of relatives for other causes, low gene penetrance, denial of the disease, non-paternity.22 Third, family size influences the probability of identifying other affected members23 with members of small families having a higher probability of appearing sporadic, particularly when related to medium-low penetrance gene mutations. Fourth, relatives of apparently sporadic ALS patients have an eightfold increased risk of developing ALS compared with the general population.24 Fifth, studies on twins have shown that hereditability for ALS accounts for up to 60%, while environmental factors account for the remaining 40%.25 Sixth, the possibility of de novo mutations, occasionally demonstrated with FUS and less commonly with SOD1.26 ,27
Clinical boundaries of ALS and their relation to ALS hereditability
The recent discovery of C9ORF72 expansions as the most relevant mutation involved in both ALS and FTD, which can manifest together in the same patient or coexist in the family, has complicated the definition of ‘positive family history’.3 ,4
Clinically incomplete forms of motor neuron disease, that is, progressive muscular atrophy (PMA) or primary lateral sclerosis (PLS), are not included in the revised El Escorial classification, which has been conceived to be used for the selection of patients for clinical trials.7 However, cases with pure PLS have been described to occur within familial ALS pedigrees,28 and patients with apparently sporadic PMA carry mutations of ALS-related genes with a frequency similar to sporadic ALS cases, demonstrating a genetic overlap between the three phenotypes29; this aspect has been acknowledged by the revised El Escorial classification with the inclusion of the category of ‘Clinically definite familial–laboratory supported ALS’.7
Psychological, social and ethical consequences of genetic testing
The identification of a causal mutation in ALS patients has profound effects on their family, since all biologic members may be at risk of having inherited the mutation. The whole family may experience emotional, psychological, ethical and legal implications.17 Psychological effects involve uncertainties about the predictivity of genetic testing, sometimes uninformative, for the positive subject, concerns about relatives and different attitudes toward sharing genetic information with family members. Motivations to pursue a test are various and include the wish to reduce anxiety, future and reproductive planning, lifestyle changes,6 altruism in contributing to research and acquisition of knowledge for the benefit of offspring and relatives.30 Reasons to refuse a predictive test include fear of the results, inability to face the risk of developing the disease, wish to maintain hope, desire to spare relatives worry if tested positive.6 Moreover, subjects who are negative to a predictive test may experience a feeling of isolation and estrangement up to a sense of guilt toward their positive relatives (known as survivor guilt).31
Disclosure of genetic test results to family members
The issue of disclosure of genetic knowledge to family members will increasingly involve ALS patients and professionals. Studies in oncology have shown that most mutation carriers are eager to share the information, and often reconnect with distant family members. Some, however, prefer not to disclose their results due to estranged family relationships, unresolved conflicts, perceived or actual vulnerability and receptivity of relatives, or uncertainties about whether relatives would wish to be informed.32 Professional ethics codes support the notion that health professionals’ obligation is toward the patient, unless there is a risk of serious harm to others, and concur that they should respect their patients’ decisions with regard to disclosure.33 Though no ‘Duty to Warn’ applies to genetic testing,34 physicians caring for ALS patients may be caught in a quandary, especially when caring for more family members at once.35 They will require training and support in discussing these issues with their patients and responding to relatives’ questions.36 The American Society of Clinical Oncology, for example, urges oncologists to clearly explain the nature of the genetic information and its potential relevance to patients' families, while refraining from being judgmental, through repeat encounters and proper counselling referrals.33
Ethical and legal implications of genetic testing
The ethical and legal implications of genetic testing include: information and informed consent, right to be tested or not, rights of others, patients’ and professionals’ responsibility to share results with at-risk relatives, confidentiality and privacy, risk of discrimination, consequences of prenatal diagnosis, and samples’ property and disposal. These issues have been widely studied in the context of cancer susceptibility and continue to be analysed as genetic testing becomes increasingly common and complex, and is often unrelated to available preventive or treatment measures.37–39
Genetic discrimination against asymptomatic individuals or their relatives on the basis of actual or presumed genetic differences or characteristics has occurred in recent history.40 Mutation carriers can be subject to different forms and degrees of discrimination in life or health insurance, at the workplace, in the process of adoption or in seeking higher education. They may also experience more subtle forms of estrangement and rejection by their families or communities, being perceived as less-than-ideal partners or parents. Similar concerns have also been reported among genetic experts and oncologists.41 Issues of genetic privacy and discrimination have been widely addressed in their ethical aspects at national and international level through regulatory measures and legislation.42 The Genetic Information Nondiscrimination Act (GINA) in the USA and the European regulation are examples of effective regulation applicable to ALS mutation carriers.43 ,44
An algorithm for genetic testing: clinical relevance and availability of an effective treatment
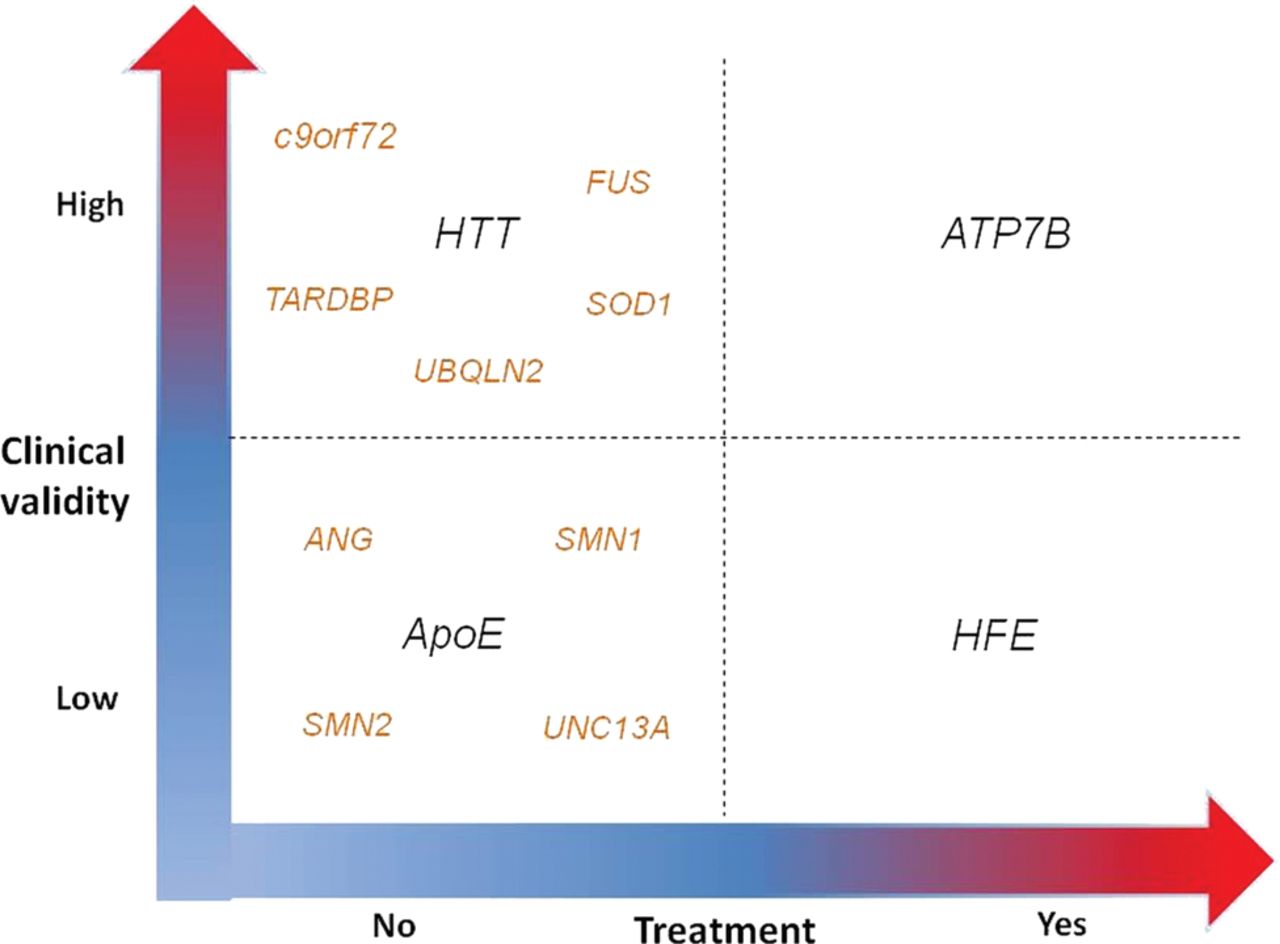
In the clinical setting, genetic testing may be categorised according to clinical relevance and availability of an effective treatment.45 Clinical relevance is related to the test reliability to predict a high risk for the disease, therefore including the concept of high penetrance. An example of high clinical relevance is Huntingtin (HTT) for HD, while of low clinical validity is ApoE for Alzheimer disease. Availability of an effective treatment refers to treatments that can cure, improve or prevent a disease, as in phenylketonuria or Wilson's disease (ATP7B gene).
Today, all ALS-related genes are characterised by the absence of an effective treatment or preventive measures, although some of them have a relatively high clinical relevance (figure 1). Therefore, for asymptomatic at-risk relatives of ALS patients, the absence of both dimensions does not justify the genetic test on either medical and social grounds. On the contrary, in subjects already affected by ALS, genetic testing is justified as a support to the clinical diagnosis and, to a lesser degree, to prognosis.7
{kind=link}
Algorithm for genetic testing based on the two-dimensional clinical relevance and availability of an effective treatment.44 When referring to asymptomatic at-risk subjects, major ALS-related genes (C9ORF72, SOD1, TARDBP, FUS) have a good clinical validity but no treatment. Susceptibility genes (ATXN2, UNC13A, ANG, SMN1, SMN2) have a low clinical validity and no treatment. AND, angiogenin; ApoE, apolipoprotein E; ATP7B, ATPase Cu transporting beta polypeptide; ATXN2, ataxin 2; C9ORF72, chromosome 9 open reading frame 72; FUS, fused in sarcoma; HFE, high iron fe; HTT, Huntingtin; SMN1, survival motor neuron 1; SMN2, survival motor neuron 2; SOD1, superoxide dismutase 1; TARDBP, transactive response-DNA binding protein.
Clinical suggestions for genetic counselling and testing
It should be understood and discussed with all patients that, once genetic testing is performed in one member (patient or relative), the entire biological family is inevitably involved in the genetic knowledge derived from the test of the index case, while not all patients or at-risk relatives may wish to be tested or to know the results of genetic testing. The ‘Right Not to Know’ is a necessary corollary of the principle of autonomy and a basic principle of human clinical genetics recognised by most international regulatory statements and legislation.30 ,44 It should be noted, however, that in the ethical debate surrounding genetic testing some authors claim that such a right amounts to a ‘Right to Ignorance’ and is not an expression of true autonomy. They rather invoke the individual's responsibility to seek and share genetic information in view of family and community ties. As the issues of autonomy, right not to know, privacy and duty to warn are now being revisited by authoritative sources, such as the American College of Medical Genetics and Genomics in its latest recommendations of March 2013, and—to a lesser extent—by medical specialty societies, also neurologists and geneticists involved in testing for ALS should become familiar with the evolution of the ethical debate in clinical genetics in order to provide appropriate counselling to their patients and family members.38 ,39 ,46–50
Before proposing genetic testing, it is mandatory to obtain in all ALS patients a detailed three-generation pedigree that captures the presence of ALS, FTD, other dementias, Parkinsonism and psychiatric conditions. This should include age at disease onset, diagnosis and age at death. Medical records and, if available, autopsy studies, are essential to clarify diagnoses. Patients should be allowed to have an exhaustive discussion, with a multidisciplinary team or an expert neurologist, about pros and cons of the testing before giving their consent.
Who should request the genetic testing?
Genetic testing for symptomatic ALS patients should be requested by a neurologist willing to take the responsibility to interpret and communicate the results and their meaning to the tested subject. Otherwise, the subject should be referred to dedicated or referral centres with expertise in ALS for the testing process. Genetic testing in asymptomatic at-risk subjects should be requested by a neurologist in collaboration with a clinical geneticist.
Who should undergo the genetic test?
All ALS patients who have a first-degree or second-degree relative with ALS, FTD or both, should be offered genetic testing. Since at least 5–10% of apparently sporadic ALS patients carry a mutation in known ALS genes and more are likely to be identified in the future, genetic testing should be discussed also with all other ALS patients, emphasising its still weak clinical significance and major uncertainties. These discussions will empower patients and family members to make informed decisions about whether or not to be tested or to enrol in clinical research protocols. At present, however, we do not recommend offering genetic testing to sporadic ALS patients, outside research protocols.
Genetic testing at present is not indicated in asymptomatic at-risk subjects and, therefore, should not be proposed. When requested by the subjects themselves, it should be performed only within the framework of a standardised protocol for asymptomatic at-risk testing in ALS expert centres. Genetic testing can be performed in asymptomatic at-risk subjects for research purposes, strictly following ethical guidelines for informed consent and pre-test and post-test counselling.2
Careful consideration is needed when the asymptomatic at-risk family members are children or adolescents. The recommendations of the European Society of Human Genetics51 state that ‘asymptomatic at risk genetic testing of minors for conditions with adult-onset is acceptable only if preventive actions (eg, therapeutic interventions) can be initiated before adulthood’. Therefore, at present, we absolutely discourage parents from requiring ALS genetic analysis for their children, and recommend that the reasons for this conduct be explained, that is, that genetic testing removes the child's ability to make an informed decision about testing when they reach adulthood, and it carries major potential psychosocial implications.52 ,53 Similar considerations apply to preimplantation or prenatal genetic testing for ALS, which are technically feasible as demonstrated by anecdotal reports.54 ,55 Finally, as the clinical significance of genetic testing for disease predisposition, including ALS, will increase, the ethical and psychosocial ramifications of testing incompetent subjects will need to be reconsidered.39 ,56
Why is the test requested?
For symptomatic subjects
At present, the main reason to request a genetic test in symptomatic patients is to provide a diagnostic support. When possible, the detected mutation can be also used for prognostic purposes and for future potential targeted therapeutic and/or preventive measures. Genetic information will also help to elucidate molecular mechanisms underlying the disease.
For asymptomatic at-risk subjects
The usefulness of genetic testing for asymptomatic at-risk subjects in the clinical setting is presently undetermined and, therefore, related to future developments. Asymptomatic at-risk testing can be proposed for research studies, provided that all relevant ethical issues are considered and standards correctly applied.
How is the test offered and presented to the patient (pre-test discussion)?
After a diagnosis of ALS is established, a discussion about ALS genetics should be introduced in person, and meanings and limitations of genetic tests explained in detail. A follow-up appointment should be offered for further questions. Patients will receive an information leaflet only after a thorough discussion.
Informed consent
The informed consent must be exhaustive and include specific statements related to present and future use of samples. In many countries, long-term storage of DNA samples is forbidden by legislation, and using clinical samples for research purposes is generally not accepted both in the USA and Europe. In some EU countries, long-term storage for clinical purposes is allowed when clearly stated in the informed consent.44 Given the lack of clear and uniform legislation on the difference between samples for research and clinical purposes, and on disclosure of genetic information, balancing privacy issues with potential advantages and drawbacks of sharing clinical and research genetic data with patients and their relatives, a strong partnership with patients and families is key to maximising the individual benefit of translation of genetic information, and to engage and educate research participants.39 We recommend that, when allowed by national regulation, each subject be asked to indicate in the informed consent whether he/she agrees with sample storage and its use for future genetic tests in case novel genetic mutations related to ALS will be discovered. The subject will be informed of the availability of new results and will be free to decide whether to know them or not. In all cases, the subject must indicate (1) if he/she wishes to know the results, (2) with whom results could be shared by the professional, (3) to whom results should be communicated in case of patient incapacitation or death.
The presence of comorbid FTD in ALS patients should be assessed in order to establish their decision-making capacity, particularly regarding informed consent for genetic studies.56 A cognitive dysfunction not reaching the level of full-blown dementia does not impair patients’ competence.57 In any case, national regulations about competence should be carefully applied.
Information leaflet
The information leaflet should include the following: (1) information about ALS, (2) the purpose, nature and consequences of the genetic testing, (3) risks involved in the procedure, (4) limitations of test interpretation (eg, other not yet identified Mendelian genes, genetic mutations that predispose to ALS), in particular regarding C9ORF72,17 (5) practical information on what will happen next, (6) potential harm of test, (7) potential psychosocial, ethical and legal ramifications, (8) implications for family members, (9) available resources, including those offered by the ALS centre and (10) information on lay associations and support groups. The information leaflet should be periodically revised to reflect all significant scientific and clinical advances.
Genetic testing in asymptomatic at risk subjects
If an at-risk relative of an ALS patient with a known genetic mutation asks to undergo genetic testing, this should be performed only after an accurate information process, including discussion of present uncertainties of ALS genetics. In particular, at-risk relatives should be made aware that even a positive test does not provide reliable information about a future development of the disease, age of onset, symptoms severity or progression rate.6
When a relative of an ALS patient with a positive family history without known genetic mutation asks for a genetic test, he/she should be informed that the DNA will be stored for further analyses in case of future identification of novel genes, and should declare whether he/she wants to be informed of the results. Phone numbers, mail or e-mail addresses, emergency contacts and responsible professionals should be listed for the person to be tested.
The process of genetic counselling: the role of a multidisciplinary team
A multidisciplinary team, including neurologist, geneticist and psychologist, is recommended for the management of genetic counselling for asymptomatic at-risk subjects. Pretest psychological assessment should be performed through: (a) psychological interview(s), focused mainly on motivations, expectations and family dynamics; (b) specific psychological and neuropsychological tests including assessment for personality, depression, illness representation, suicidal tendency and cognitive function.
Where and how is the test performed?
Genetic testing should be performed in accredited medical genetics laboratories according to international quality standards.58 Whenever possible, for each positive sample, a second test on a new blood sample is recommended.
Both in-house developed and commercial assays should be validated before use. The identification of point mutation and small del/ins is performed with Sanger sequencing method. For expansions detection the Repeat Primed PCR (RP-PCR) protocol is followed.16
What genes should be tested?
According to the available literature which mostly refers to population of Caucasian or East Asian ancestry, two-thirds of mutations are found in four genes, C9ORF72, SOD1, TARDBP and FUS. Therefore, these genes should be considered for a routine diagnostic protocol. C9ORF72 testing is worthwhile in sporadic patients as this gene has been shown to be present in a significant proportion of such cases, as well as in ALS patients with relatives affected by FTD, while the other genes are more rarely found in sporadic cases. If these four genes are negative, second-level genetic analysis can be performed, assessing other ALS-related genes, according to the country-specific genetic epidemiology when available. UBQLN2 should be tested in presence of a suspected X-linked dominant inheritance. The list of genes should be periodically modified along with increasing knowledge of ALS genetics.
In order to guide the genetic laboratory, the following demographic and clinical information should be provided by the proponent physician: (1) gender, (2) age and site of onset, (3) comorbidities (in particular FTD and extrapyramidal features), (4) other members affected by ALS or FTD and their relationship with the index case, (5) ethnicity, (6) place of birth and area of origin of parents and grandparents. This information is very important to avoid the risk of inadvertently performing an asymptomatic at-risk test outside the proper counselling context and procedures.
Although some genes have been proposed as ALS modifiers (ie, ATXN2, UNC13A, SMN1 and SMN2), these should not be performed for clinical purposes, but reserved only for the research setting.
What information should be included in the genetic report?
The report should contain demographic information, detailed list of the genes tested and the exons analysed for each gene, test methodologies, limitations of each methodology (false positive and negative percentage) and explanation of the genetic findings. Unreported genetic variants should be discussed in order to distinguish, when possible, mutations from non-pathogenetic polymorphisms.
How are the results handled by the proponent professional (post-test discussions)?
The results of the genetic test, either positive or negative, should be communicated in person to the patient by the proponent professional, and never by letter, e-mail or phone. Ideally, discussions of genetic test results with asymptomatic at-risk subjects should take place in a team context, with the concomitant or subsequent presence of a geneticist and a psychologist. As this may be unfeasible in many clinical settings, we recommend as minimal requirement that test result discussions be performed by a neurologist skilled in ALS genetics. All tested subjects are encouraged to be accompanied by a trusted family member or friend to help support them in the processing of the information received. The patient should be given time to process the information and ask questions, and a follow-up appointment should be organised for further questions. Psychological support should be offered to the patient tested and to at-risk relatives informed by the patient.
Confidentiality
Results of genetic testing are highly sensitive, and rigorous measures to ensure confidentiality are necessary. Results must never be disclosed to a third party without explicit written consent from the patient or their lawful surrogates. To avoid unintentional disclosure, test results must be kept in a separate file and not in the patient's medical records.2 However, this measure could be modified over time, when genetic testing will be better understood in its clinical meaning and limitations rather than in a deterministic way that equates genetic knowledge with the notions of ‘future diaries and destiny’.38 ,59 Also, gene therapy for many diseases, including ALS, may become available in the future, and withholding information could prevent patients and/or family members from receiving effective treatment measures.
Conclusions
Genetic information about ALS is rapidly growing but not yet fully translated into clinically useful knowledge. While we could be tempted to refrain from discussing with our patients about information that is already exceeding our comfort level as professionals, we must, however, consider the potential benefits of genetic information for patients’ autonomy. Despite major cultural, social and religious differences among different populations that prevent generalisations about attitudes and practices related to genetic testing and the universal applicability of clinical recommendations, the communication process that accompanies and follows genetic testing for ALS should be standardised following existing clinical experiences and guidelines in neurologic and other diseases. Further research in clinical and epidemiological genetics of ALS is urgently needed to improve the process of genetic counselling for our patients (box 1). The aim of our clinical suggestions is to enable neurologists and ALS specialists to provide optimal multidisciplinary clinical consultations to our patients and families.
Priority areas for amyotrophic lateral sclerosis (ALS) research in clinical genetics and genetic counselling
A. ALS clinical genetics
▸ Identification of the pathogenicity of different ALS genes and, in each gene, of different mutations.
▸ Identification of penetrance of different genes/mutations.
▸ Identification of cut-off size of pathogenic GGGGCC expansion in C9ORF72 gene.
▸ Identification of genetic-phenotypic correlations of different ALS genes/mutations.
▸ Identification of frequency of ALS-related genes/mutations in various populations.
B. Genetic counselling in ALS
▸ Identification of psychological, social and ethical factors implicated in genetic counselling of ALS patients and their relatives, including asymptomatic at-risk subjects.
▸ Quality assessment of genetic counselling process in the context of ALS patients and presymptomatic subjects testing.
In the future, we may increasingly be asked to consult on genetic tests recommended or prescribed by other physicians, or obtained through direct-to-consumer tests. Requests for testing asymptomatic minors, and for prenatal diagnosis, are also likely to increase. It is therefore our professional duty as ALS experts to be constantly updated about the developments in our field, to help refining and updating clinical suggestions and recommendations based on new genetic knowledge, and to promote continuous medical education in all its medical and communicative aspects.
Acknowledgments
The Corteranzo meeting was supported by funds of the Department of Neuroscience, University of Torino, Italy. A Chio received funding from the Health Seventh Framework Programme (FP7/2007–2013) under grant agreement number 259867; the Italian Ministry of Health (grant RF-2010-2309849); Federazione Italiana Giuoco Calcio; and Agenzia Italiana Ricerca per la SLA (grant 2012). MS received funding from Federazione Italiana Giuoco Calcio. GM received funding from the Italian Ministry of Health (grant RF-2010-2309849). MC received funding from the Italian Ministry of Health (grant RF-2010-2309849).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online tables
Footnotes
-
Collaborators List of the others members of the ITALSGEN Consortium. Maria Rosaria Monsurrò, Gioacchino Tedeschi (Naples); Amelia Conte, Marco Luigetti, Serena Lattante, Giuseppe Marangi (Rome); Paolo Volanti (Mistretta); Kalliopi Marinou, Laura Papetti, Christian Lunetta, Giuseppe Lauria Pintor (Milan); Fabrizio Salvi, Ilaria Bartolomei (Bologna); Aldo Quattrone, Antonio Gambardella (Catanzaro); Giancarlo Logroscino, Isabella Simone (Bari); Fabrizio Pisano (Veruno); Rossella Spataro, Vincenzo La Bella, Tiziana Colletti (Palermo); Gianluigi Mancardi, Paola Origone (Genoa); Patrizia Sola (Modena), Giuseppe Borghero, Francesco Marrosu, Maria Giovanna Marrosu, Maria Rita Murru; Gianluca Floris, Antonino Cannas, Valeria Piras, Emanuela Costantino, Carla Pani (Cagliari); Maria Alessandra Sotgiu, Maura Pugliatti, Leslie D. Parish, Paola Cossu (Sassari); Anna Ticca (Nuoro); Carmelo Rodolico, Simona Portaro (Messina); Claudia Ricci (Siena); Cristina Moglia, Irene Ossola, Maura Brunetti, Marco Barberis, Antonio Canosa, Stefania Cammarosano, Davide Bertuzzo, Giuseppe Fuda, Antonio Ilardi, Umberto Manera (Torino); Ilaria Pastore (Cuneo); William Sproviero (Cosenza); Francesco Logullo (Ancona); Raffaella Tanel (Trento).
-
Contributors A group of researchers with different expertise and focused on ALS genetics met at Corteranzo, Italy, to debate controversies in ALS genetics in order to establish recommendations for the process of genetic counselling in ALS patients and asymptomatic at risk subjects. A Chio and GR conceived and planned the Corteranzo meeting. All authors contributed to meeting discussions. AC and GM provided the initial drafts and figures. All authors contributed to the editing of the manuscript, with final editing undertaken by A Chio, GM, GR, AS and MS.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial commentary
- Editorial commentary