Article Text

Abstract
Background Clinical trials established the efficacy and safety of natalizumab. Data are needed over longer periods of time and in the clinical practice setting.
Objective To evaluate long-term safety of natalizumab and its impact on annualised relapse rate and Expanded Disability Status Scale (EDSS) progression in patients with relapsing-remitting multiple sclerosis (RRMS).
Methods The Tysabri (natalizumab) Observational Program (TOP) is an open-label, multinational, 10-year prospective study in clinical practice settings.
Results In this 5-year interim analysis, 4821 patients were enrolled. Follow-up for at least 4 years from natalizumab commencement in 468 patients and at least 2 years in 2496 patients revealed no new safety signals. There were 18 cases of progressive multifocal leucoencephalopathy reported, following 11–44 natalizumab infusions. Mean annualised relapse rate decreased from 1.99 in the 12 months prior to baseline to 0.31 on natalizumab therapy (p<0.0001), remaining low at 5 years. Lower annualised relapse rates were observed in patients who used natalizumab as first MS therapy, in patients with lower baseline EDSS scores, and in patients with lower prenatalizumab relapse rates. Mean EDSS scores remained unchanged up to 5 years.
Conclusions Interim TOP data confirm natalizumab's overall safety profile and the low relapse rate and stabilised disability levels in natalizumab-treated patients with RRMS in clinical practice.
Trial registration number NCT00493298.
- Multiple Sclerosis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Natalizumab (Tysabri, Biogen Idec Inc, Cambridge, Massachusetts, USA) is a selective adhesion molecule inhibitor that blocks α4 integrin, which is expressed on the surface of lymphocytes and is required for endothelial adhesion, facilitating migration of peripheral blood lymphocytes into the central nervous system.1
In the 2-year, phase 3 AFFIRM study2 of patients with relapsing-remitting multiple sclerosis (RRMS), natalizumab monotherapy demonstrated consistent efficacy in the overall study population and across multiple subgroups of patients predefined on the basis of demographic and baseline disease characteristics, including age, sex, number of brain MRI lesions, disability status and number of relapses in the prior year.2 ,3
While the AFFIRM trial established the safety and efficacy of natalizumab, data are needed to confirm the safety and efficacy of natalizumab over treatment durations longer than 2 years and, importantly, in a clinical practice setting.
The Tysabri (natalizumab) Observational Program (TOP) was designed to evaluate the long-term safety of natalizumab monotherapy, as well as its impact on disease activity and disability progression, in patients with RRMS in the clinical practice setting. This paper presents findings from an interim analysis of TOP data from study initiation in July 2007 to a data lock on 1 December 2012.
Methods
Patients
Eligible RRMS patients met criteria for natalizumab prescription in their respective countries and had three or fewer natalizumab infusions before enrolment in TOP. Female participants were postmenopausal, surgically sterile, or willing to practice effective contraception.
Patients were enrolled as soon as possible after deciding to start natalizumab treatment. The latest permissible enrolment date was the day before patients’ fourth natalizumab infusion (ie, 16 weeks after the start of natalizumab treatment).
The protocol was approved by each centre's independent ethics committee. The study was performed in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. All patients provided written informed consent.
Study design
TOP is an ongoing, open-label, multinational, multicentre, prospective, observational study conducted in clinical practice settings in Europe, Australia, Canada and Argentina. Patients receive natalizumab 300 mg intravenously over approximately 1 h, every 4 weeks.
Endpoints and assessments
Data were collected at regular clinical visits every 6 months. Data entry was Web based. Patients who discontinued natalizumab were encouraged to remain in the study; if patients exited the study for any reason, physicians were asked to collect data on serious adverse events for an additional 6 months where possible.
The primary endpoint is long-term safety (incidence and type of serious adverse events). Secondary endpoints include measures of MS disease activity (ie, the occurrence of clinical relapses) and change in Expanded Disability Status Scale (EDSS) score. Multiple Sclerosis Severity Scale4 scores were recorded at baseline.
A clinical relapse was defined as new or recurrent neurological symptoms, not associated with fever, lasting for ≥24 h and followed by a period of 30 days of stability or improvement. New or recurrent neurological symptoms that occurred <30 days following the onset of a protocol-defined relapse were considered part of the same relapse.
Confirmed EDSS progression was defined per protocol as an increase of ≥1.0 point in EDSS score sustained for 6 months. We also performed analyses comparable to those used in recent clinical trials, in which confirmed EDSS progression was defined as an increase, sustained for 6 months, of ≥0.5 points from a baseline EDSS score ≥6.0, of ≥1.0 point from a baseline EDSS score of ≥1.0 to <6.0 or of ≥1.5 points from a baseline EDSS score of 0.0.2 ,5 Confirmed EDSS improvement was assessed in the overall population and in patients with baseline EDSS scores ≥2.0 and was defined as a decrease of ≥1.0 point in EDSS score sustained for 6 months.6
Medical review of each case report was performed at the end of data entry. Serious adverse events were evaluated for possible clinical significance or indication of a previously unknown risk. The Drug Safety Department at Biogen Idec evaluated serious adverse event reports to ensure case validity and confirm seriousness criteria. The contract research organisation was notified of discrepancies for resolution with the site investigators.
Study endpoints were assessed uniformly across sites. To assure standardised examinations and consistent definitions for the EDSS Functional System (FS) scores, participating physicians were provided a copy of the interactive Neurostatus Training DVD-ROM,7 and Neurostatus certification was highly recommended. Investigators not previously certified were offered free-of-charge online certification (http://www.neurostatus.net). To reduce the risk of entry error with EDSS score reporting, the electronic case report form (CRF) calculated an EDSS score based on the Kurtzke FS and ambulation scores that were entered. The investigator either confirmed the calculated score or entered their own score with an explanation of why the proposed score was incorrect.
CRFs were designed to automatically generate queries for data inconsistencies, including data that were out of range or otherwise invalid. Data quality control procedures checked for consistency across data sets. Site-based verification and correction was used for residual data queries.
Statistical analysis
This study is designed to include at least 4500 patients. The sample size allows detection of a serious adverse event with an incidence of 1/1000 with 95% probability. Enrolment started in July 2007, and patients will be followed for up to 10 years.
Descriptive statistics were used to summarise baseline characteristics and outcome measures. Efficacy and safety analyses, without imputation of missing data, were performed using the population of patients who provided a consent form, met inclusion and exclusion criteria and had received three or fewer doses of natalizumab at enrolment.
Annualised relapse rates were estimated using a negative binomial model. Time to EDSS progression and improvement and cumulative probabilities were analysed using the Kaplan–Meier method.
A repeated negative binomial model was used for a paired comparison of annualised relapse rate prenatalizumab and postnatalizumab treatment start. Associations between baseline factors and on-treatment annualised relapse rates were evaluated using a negative binomial model. A univariate analysis of each demographic and baseline disease characteristic with on-treatment annualised relapse rates was performed. Any variable with a p value <0.25 was considered in a multivariate negative binomial model. Baseline variables included in the adjusted negative binomial model were sex, relapse, EDSS score, disease duration, number of previous disease-modifying therapies (DMTs) used and treatment duration.
To further assess associations between baseline treatment history and on-treatment annualised relapse rates, patients were stratified into five treatment-history groups comprising therapies commercially available at the start of the study: therapy naïve, interferon β-1 (IFN) only, glatiramer acetate (GA) only, exposed to IFN and GA (either order), and a final group with any prior immunosuppressant (IS) use. A negative binomial regression model was used, adjusting for the baseline covariates described above.
Associations between baseline treatment history and postbaseline serious adverse events were evaluated using Pearson χ2 or Fisher exact tests and Poisson model. For this analysis, patients were stratified into three groups: therapy naïve, at least one prior DMT without prior IS use, and prior IS use.
Results
Patient disposition and baseline characteristics
As of 1 December 2012, 4821 patients from 16 countries were enrolled in TOP. Table 1A,B summarises baseline characteristics. Differences between the AFFIRM and the TOP populations at baseline are described in table 1A. Table 2 summarises patient enrolment and disposition data. Overall, 77% (n=3703) were followed for at least 1 year, 52% (n=2496) for at least 2 years, 25% (n=1220) for at least 3 years and 10% (n=468) for at least 4 years. The median follow-up period was 26 months (range 1–69). Of the 4821 enrolled patients, all received at least one dose of natalizumab. The median duration of natalizumab therapy was 22 months (range 1–74).
Baseline characteristics
Patient enrolment and disposition
Overall, 1222 of 4821 patients (25.3%) discontinued natalizumab treatment and 740 of 4821 (15.3%) withdrew from TOP; 523 patients discontinued natalizumab and withdrew from study follow-up. Patients may have discontinued natalizumab for more than one reason; the most common reasons included anti-JC virus (JCV) antibody positive status (n=277; 5.7% of all TOP patients), medication change (n=247; 5.1%), insufficient efficacy (n=229; 4.8%) and patient decision (n=171; 3.5%). Fifty-one patients discontinued due to serious adverse events. The most common reasons for study withdrawal included loss to follow-up (n=121; 2.5%), medication change (n=116; 2.4%), patient decision (n=115; 2.4%), withdrawal of consent (n=110; 2.3%) and movement out of the area (n=99; 2.1%).
Baseline characteristics were comparable between the 1222 patients who discontinued natalizumab treatment and the 3599 patients who remained on treatment, with the exception of a higher proportion of patients with prior IS use among those who discontinued (18%) versus those who remained on therapy (13%, p<0.0001).
Safety
Table 3 summarises serious adverse events recorded at the time of data lock. Overall, 8.0% of patients experienced a serious adverse event and 2.6% experienced a serious adverse event that was considered related or possibly related to natalizumab. The most common serious adverse event was infection (incidence 1.9%). Among the 97 serious infections, there were 18 herpes infections, 17 urinary tract infections and 16 pneumonias. Twelve of the 97 serious infections were identified as opportunistic; these included two herpes zoster, two herpes meningitides and two pneumonias, as well as one each of urinary tract infection, pyelonephritis, streptococcal meningitis, staphylococcal sepsis, encephalitis and Mycobacterium kansasii infection. The incidence of serious hypersensitivity reactions was 0.5%, including anaphylaxis/anaphylactoid reactions (0.2%).
Summary of serious adverse events with an incidence >1
Eighteen patients were diagnosed with progressive multifocal leucoencephalopathy (PML) after a median of 29 natalizumab doses (range 11–44). Fourteen of the 18 PML cases occurred in patients receiving natalizumab for ≥2 years; four PML patients previously used IS (mitoxantrone in three cases; azathioprine in the fourth); 13 of the patients had received at least one DMT prior to natalizumab. The overall PML incidence rate was 0.4% (3.73/1000), with a 95% CI of 0.2 to 0.6. Seven of the 18 PML patients had confirmed positive anti-JCV antibody serostatus at least 6 months (6–15 months) prior to PML development; serostatus at least 6 months prior to PML development was unknown or not reported for 11 patients. At the time of this analysis (December 2012), 16 of the 18 patients (89%) diagnosed with PML were alive.
There were 24 patients with 12 types of malignancies. Breast cancer was the most common, affecting seven patients (all female). Papillary thyroid cancer was diagnosed in three patients; cervical cancer, melanoma (one in situ) and leukaemia were each diagnosed in two patients; the other cancer diagnoses (rectal cancer, colon cancer, glioblastoma, basal cell carcinoma, kidney tumour, prostate cancer and testicular neoplasm) each occurred in one patient.
Nine deaths (9/4821=0.2%) occurred during the study. One death was attributed to urosepsis (patient also had PML, which was ongoing at time of death), and there were three suicides, two fatal pulmonary emboli, one fatal drowning, one death due to autonomic nervous system imbalance and one death due to thermal burn.
Relapses
Overall, the annualised relapse rate (95% CI) decreased from 1.99 (1.95 to 2.03) in the 12 months prior to baseline to 0.31 (0.29 to 0.32) on natalizumab therapy (p<0.0001). Relapse rates in each year of natalizumab exposure ranged from 0.21 to 0.30 (figure 1).
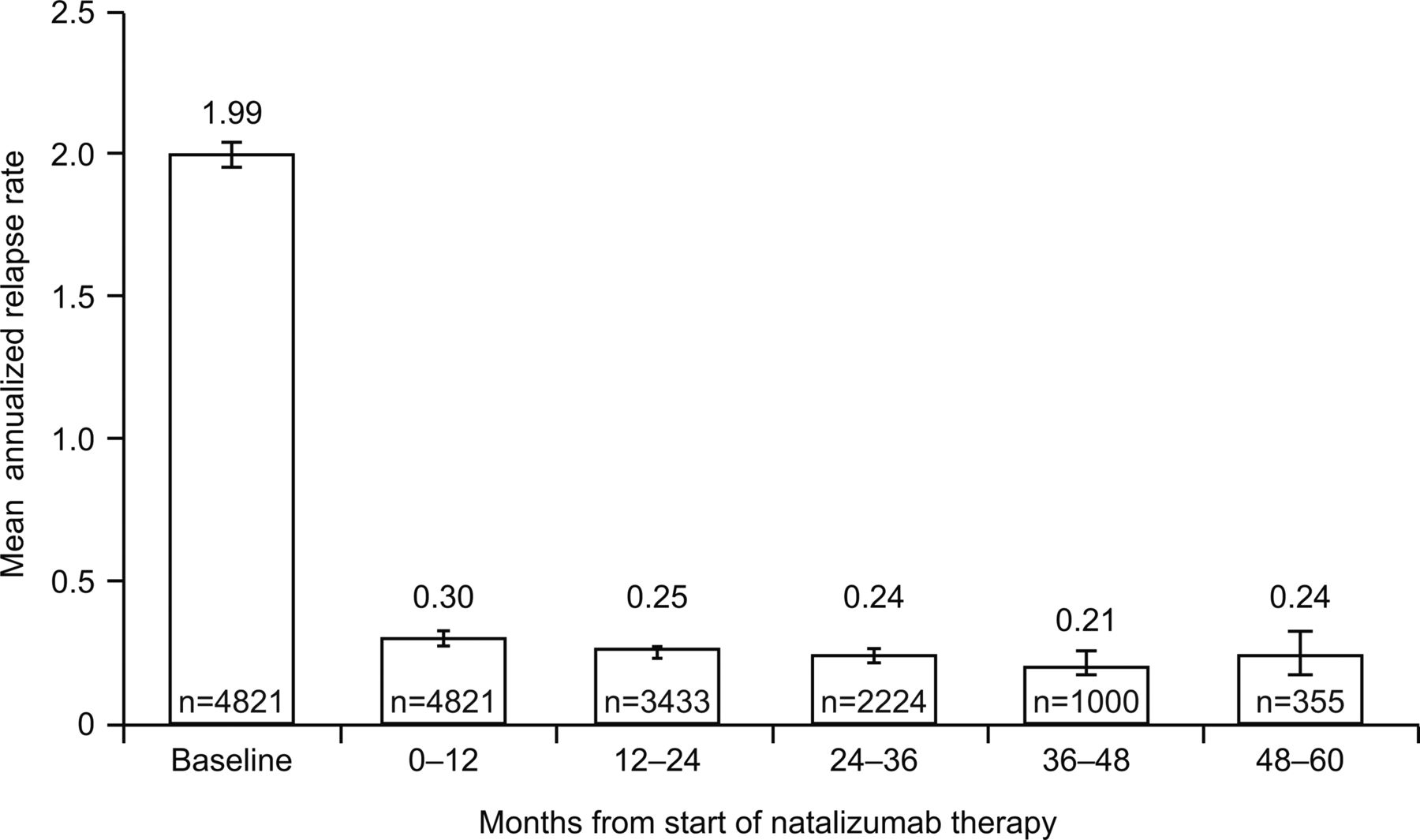
Annualised relapse rate on natalizumab therapy: per yearly interval over time (n=4821). Error bars indicate 95% CIs. Data beyond 5 years are excluded.
Significant associations between baseline disease characteristics and annualised relapse rates on natalizumab therapy were observed (figure 2). Lower mean on-therapy annualised relapse rates were associated with lower baseline EDSS scores, fewer relapses in the prior year and fewer DMTs used prior to natalizumab (figure 2A). When therapy history and baseline relapse status were analysed together, the lowest on-therapy annualised relapse rates were seen in patients with a history of only one relapse in the prior 12 months who were either therapy naïve or had received only one DMT prior to natalizumab; those with greater numbers of relapses and/or DMTs had higher rates (0.16 to 0.18 vs 0.23 to 0.40; p<0.0001) (figure 2B). While patients treated with natalizumab had significant reductions in annualised relapse rate regardless of treatment history, annualised relapse rates were lowest in patients who were therapy naïve at baseline and highest in those patients with prior IS use (figure 2C). In pairwise comparisons, annualised relapse rates were significantly different for the therapy-naïve group versus each of the other groups and for any of the comparisons of GA and/or IFN use groups versus the prior IS use group (p<0.05).

Annualised relapse rate after initiation of natalizumab according to (A) baseline characteristics, (B) combined baseline relapse status and number of prior disease-modifying therapies (DMTs) and (C) baseline treatment history.*p Value from a negative binomial regression model adjusted for sex, baseline EDSS score (<3.0 vs ≥3.0), relapse status in the past year (≤1 vs >1), prior DMT use (0, 1 and >1), disease duration (≥8 vs <8 years) and treatment duration (≥3 vs <3 years). †p=0.697 between comparator groups at baseline on the basis of a negative binominal model for baseline annualised relapse rate (adjusted for sex, baseline EDSS score (<3.0 vs ≥3.0), disease duration (<8 vs ≥8 years) and treatment duration (<3 vs ≥3 years)). ‡p<0.0001 for difference among groups after treatment is from negative binomial model adjusted for sex, baseline relapse status (0 or 1 relapse vs >1), EDSS score (<3.0 vs ≥3.0), treatment duration (<3 vs ≥3 years) and disease duration (<8 vs ≥8 years). Error bars indicate 95% CIs. DMT, disease-modifying therapy; EDSS, Expanded Disability Status Scale; GA, glatiramer acetate; IFN, interferon β-1; IS, immunosuppressant.
In the overall population, the cumulative probabilities of remaining relapse free by 1, 2, 3, 4 and 5 years on treatment were 78.9%, 67.1%, 56.3%, 50.0% and 44.4%, respectively.
The 229 patients who discontinued natalizumab due to ‘insufficient efficacy’ had more severe disease than the overall TOP population prior to natalizumab treatment, as indicated by a higher median baseline EDSS score (4.5 vs 3.5) and a higher median baseline Multiple Sclerosis Severity Scale score (6.4 vs 4.9). Compared with the overall TOP population, a greater proportion of these patients relapsed during the first year in the study (45.4% vs 18.1%). While antinatalizumab antibody status data were not collected routinely during the study, the presence of antinatalizumab antibodies was listed as a reason for discontinuing natalizumab for 44 of the 1222 patients (3.6%) who discontinued treatment.
Impairment and disability
In the overall population, the mean (SD) EDSS score was 3.5 (1.62) in 4797 patients at baseline, 3.3 (1.76; n=2064) after 1 year, 3.3 (1.84; n=1304) after 2 years, 3.3 (1.84; n=744) after 3 years and 3.3 (1.92; n=325) after 4 years of observation.
Using the prespecified definitions, the cumulative probability of confirmed EDSS progression at 4 and 5 years was 14%. The cumulative probability of confirmed EDSS improvement was 24% at 4 years and 27% at 5 years. Using the clinical trial definition of EDSS progression as defined above, the cumulative probability of confirmed EDSS progression at 5 years was 16% (figure 3A). The cumulative probability of improvement at 5 years in the subgroup of patients with baseline EDSS scores ≥2.06 was 29% (figure 3B). The probability of confirmed EDSS improvement was significantly higher than the probability of confirmed worsening (p<0.0001).

{kind=link}
{kind=link}
{kind=link}
(A) Time from first natalizumab infusion to confirmed EDSS progression in overall study population. (B) Time from first natalizumab infusion to confirmed EDSS improvement in patients with baseline EDSS scores ≥2.0. EDSS progression was defined as an increase, sustained for 6 months, of ≥0.5 points from a baseline EDSS score ≥6.0, of ≥1.0 point from a baseline EDSS score of ≥1.0 to <6.0 or of ≥1.5 points from a baseline EDSS score of 0.0. EDSS improvement was defined as a decrease, sustained for 6 months, of ≥1.0 point from baseline EDSS score. EDSS, Expanded Disability Status Scale.
It is noteworthy to emphasise that the rate of 12-month confirmed EDSS progression, which might be informative for secondary progression, was not significantly different between the 229 patients who discontinued treatment due to ‘insufficient efficacy’ (median follow-up 13 months) as the stated reason for natalizumab cessation and the 3580 patients who remained on treatment (median follow-up 23 months): 6% and 4% of patients, respectively, had confirmed EDSS progression (p=0.118).
Discussion
In this interim analysis of the first 5 years of the TOP study, safety findings were consistent with previous studies of natalizumab,2 ,5 ,8–10 and no new safety concerns were identified in a clinical practice setting. The most frequently reported serious adverse event in TOP to date was infection (1.9%), which was also the most frequently reported serious adverse event (aside from MS relapse) among all patients receiving natalizumab in the phase 3 monotherapy trial (3.2%).2 ,11 There were 17 reported cases of serious urinary tract infection (0.4%) and 16 serious pneumonias (0.3%) in TOP, compared with rates of 0.8% and 0.6%, respectively, in natalizumab-treated patients in the phase 3 trial.2 ,11 Opportunistic infections other than PML were reported with an incidence of 0.2% in TOP, which is consistent with the rate of <1% observed in clinical trials.2 ,11
The overall PML incidence in TOP (0.4% (3.73 cases/1000 patients)) is similar to the most recently reported risk in the natalizumab postmarketing setting (3.40 cases/1000 patients).12 Fourteen of 18 PML cases occurred in patients receiving natalizumab for ≥2 years. In all seven cases with reported anti-JCV antibody serostatus at least 6 months prior to PML development, results were positive. These data are consistent with the known markedly increased risk of PML in anti-JCV antibody positive patients with a longer duration of natalizumab therapy.13 The risk of PML in patients who are anti-JCV antibody positive, have had prior IS use and have been exposed to natalizumab for 25–48 months is estimated at 11.2 cases per 1000.12 The risk in patients who are anti-JCV antibody negative is estimated at 0.1 cases per 100012; in these patients, prior IS use and treatment duration do not affect PML risk as long as the patients remain anti-JCV antibody negative. Direct comparison of these risk estimates with those derived from TOP data cannot be made as anti-JCV antibody serostatus data were not collected throughout the protocol period. A recent protocol amendment has been approved to collect this information.
The observation that a significantly greater proportion of patients who discontinued natalizumab therapy had prior IS use compared with patients who remained on therapy suggests that decisions regarding treatment discontinuation were driven, at least in part, by PML risk factor assessment.
Rates of serious hypersensitivity reactions (0.5%) and anaphylaxis or anaphylactoid reactions (0.2%) in TOP were lower than the rates of 1.3% and 0.8%, respectively, among natalizumab-treated clinical trial patients.2
Malignancies were described in 24 patients; breast cancer was the most common neoplasm, occurring at a rate consistent with the expected European incidence rate.14 Further follow-up in TOP will evaluate the risk of malignancy with longer natalizumab exposure.
On-treatment annualised relapse rate was significantly reduced and remained low after 5 years of natalizumab exposure. EDSS scores remained stable over time, and the probability of confirmed EDSS improvement was significantly higher than the probability of confirmed worsening (p<0.0001). Even severely affected patients at baseline, namely patients with higher numbers of relapses, high EDSS scores and/or use of multiple prior therapies, achieved excellent clinical disease control, with an on-therapy annualised relapse rate ≤0.38 for all subgroups. These findings accord with other observational studies and registries, in which significant reductions in annualised relapse rates were observed over 1–3 years of natalizumab treatment in clinical practice settings.15–17
In conclusion, this interim 5-year analysis of TOP data extends findings of randomised controlled pivotal trials by describing serious adverse event incidence in a large cohort of natalizumab-exposed patients with RRMS and demonstrates the low relapse probability and low confirmed EDSS progression events associated with natalizumab treatment. Unlike the AFFIRM population, in which the majority of patients were treatment naïve,2 the majority of patients in TOP (≈90%) were previously treated with a DMT and/or IS therapy. Although relapse rates remained low across all groups on natalizumab, patients who started natalizumab treatment when they were therapy naïve, or with lower baseline EDSS scores or relapse rates, or who used fewer prior DMTs had lower on-therapy annualised relapse rates, suggesting there may be a lower clinical disease activity level when natalizumab treatment is initiated earlier in the disease course. TOP and other observational registries of natalizumab-treated patients will continue to generate valuable data on the long-term (5–10-year) safety and efficacy profile of natalizumab in a clinical practice setting.
Acknowledgments
The authors would like to acknowledge Amy Pace, ScD, of Biogen Idec Inc for her expert assistance with statistical analysis and interpretation; Dominic Paes, PhD, of Biogen Idec Inc, and Daniel Desgrandchamps, MD, formerly of Biogen Idec Inc, for their significant contributions to the study design, the start of the study and coordination of the initial 2 years of the study on behalf of Biogen Idec Inc; and Samir Mechati of Rodanotech Sarl for his contributions to database design and customisation. Katherine Hauswirth, RN, MSN, from Infusion Communications wrote the first draft of the manuscript based on input from authors, and Jackie Cannon from Infusion Communications copyedited and styled the manuscript per journal requirements.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Collaborators HB, LK, FP, MT and HW are TOP Steering Committee members (a full list of the Investigators can be found online as supplementary material).
-
Contributors HB contributed to the study design, patient recruitment, data collection, analysis and interpretation of data, and drafting and revising of the manuscript; he takes full responsibility for the finished article, access to any data, and controlled the decision to publish. LK contributed to the study design, analysis and interpretation of data, and drafting and revising of the manuscript. FP contributed to the analysis and interpretation of data and drafting and revising of the manuscript. MT contributed to the study design, patient recruitment, data collection, analysis and interpretation of data, and drafting and revising of the manuscript. HW contributed to the funding, study design, analysis and interpretation of data, and drafting and revising of the manuscript. RNP contributed to the analysis and interpretation of data and drafting and revising of the manuscript. AZ contributed to the statistical analysis, analysis and interpretation of data, and drafting and revising of the manuscript. CH contributed to the study design, analysis and interpretation of data, and drafting and revising of the manuscript. SB contributed to the study design, patient recruitment, data collection, statistical analysis, analysis and interpretation of data, and drafting and revising of the manuscript.
-
Funding This study was supported by Biogen Idec Inc, which also provided funding for editorial support in the development of this manuscript. Biogen Idec reviewed the manuscript and provided feedback on the manuscript. The authors had full editorial control of the manuscript and provided their final approval of all content.
-
Competing interests HB has been a member of scientific advisory boards for Biogen Idec, Merck Serono, Novartis and sanofi-aventis; has received conference travel support from Biogen Idec, Merck Serono, Novartis and sanofi-aventis; has received fees for steering committees for trials conducted by Biogen Idec, Merck Serono and Novartis; has received awards and grants including the National Health and Medical Research Council (NHMRC) Career Development Award, NHMRC Project Grants, NHMRC Centre of Excellence Award, Australian Research Council Linkage Grant and National MS Society (USA) Project Grant; is on the editorial board of Multiple Sclerosis International and Multiple Sclerosis and Related Disorders; and has received research support from Biogen Idec, Merck Serono and Novartis, as honorary chair of the MSBase Foundation. LK has received research support from Acorda, Actelion, Allozyne, BaroFold, Bayer HealthCare, Bayer Schering, Bayhill, Biogen Idec, Boehringer Ingelheim, Elan, Genmab, Glenmark, GlaxoSmithKline, Merck Serono, MediciNova, Novartis, sanofi-aventis, Santhera, Shire, Roche, Teva, UCB, Wyeth, Swiss MS Society, Swiss National Research Foundation, European Union, Gianni Rubatto Foundation, and the Novartis and Roche Research Foundations. FP has received consulting fees and honoraria from Biogen Idec. MT has been a consultant for and received speaker honoraria from Bayer Schering, Biogen Idec, Novartis and sanofi-aventis and has received research grants from Biogen Idec and Merck Serono. HW has received honoraria from Bayer, Biogen Idec/Elan, Medac, Merck Serono, Novo Nordisk, sanofi-aventis, Schering and Teva; has been a consultant for Bayer Vital/Schering, Biogen Idec, Medac, Merck Serono, Novartis, Novo Nordisk, sanofi-aventis and Teva; and has received research support from Bayer, Biogen Idec/Elan, Medac, Merck Serono, Novo Nordisk, sanofi-aventis, Schering and Teva. RNP, AZ, CH and SB are employees of Biogen Idec.
-
Ethics approval Ethics committees for all participating study centres.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial commentary