Article Text

Abstract
Objective To assess the efficacy of recombinant human erythropoietin (rhEPO) in amyotrophic lateral sclerosis (ALS).
Methods Patients with probable laboratory-supported, probable or definite ALS were enrolled by 25 Italian centres and randomly assigned (1:1) to receive intravenous rhEPO 40 000 IU or placebo fortnightly as add-on treatment to riluzole 100 mg daily for 12 months. The primary composite outcome was survival, tracheotomy or >23 h non-invasive ventilation (NIV). Secondary outcomes were ALSFRS-R, slow vital capacity (sVC) and quality of life (ALSAQ-40) decline. Tolerability was evaluated analysing adverse events (AEs) causing withdrawal. The randomisation sequence was computer-generated by blocks, stratified by centre, disease severity (ALSFRS-R cut-off score of 33) and onset (spinal or bulbar). The main outcome analysis was performed in all randomised patients and by intention-to-treat for the entire population and patients stratified by severity and onset. The study is registered, EudraCT 2009-016066-91.
Results We randomly assigned 208 patients, of whom 5 (1 rhEPO and 4 placebo) withdrew consent and 3 (placebo) became ineligible (retinal thrombosis, respiratory insufficiency, SOD1 mutation) before receiving treatment; 103 receiving rhEPO and 97 placebo were eligible for analysis. At 12 months, the annualised rate of death (rhEPO 0.11, 95% CI 0.06 to 0.20; placebo: 0.08, CI 0.04 to 0.17), tracheotomy or >23 h NIV (rhEPO 0.16, CI 0.10 to 0.27; placebo 0.18, CI 0.11 to 0.30) did not differ between groups, also after stratification by onset and ALSFRS-R at baseline. Withdrawal due to AE was 16.5% in rhEPO and 8.3% in placebo. No differences were found for secondary outcomes.
Conclusions RhEPO 40 000 IU fortnightly did not change the course of ALS.
- ALS
- MOTOR NEURON DISEASE
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Several in vitro and in vivo models of degenerative, toxic and inflammatory peripheral and central nervous system diseases have shown that erythropoietin (EPO) and its non-erythropoietic derivatives have neuroprotective properties.1 EPO is a circulating glycoprotein whose principal function is the production of red blood cells through the inhibition of erythroid progenitor apoptosis and regulation of differentiation in the bone marrow. Endogenous EPO and the recombinant human EPO (rhEPO) form are identical regarding the sequence of amino acids, but have heterogeneous bioavailability and pharmacokinetics due to the different composition of the sugar side chains, indicated by a Greek letter suffix (eg, α, β, γ and δ). EPO and its classical receptor (EPOR) are expressed in human neurons and astrocytes. Preclinical studies demonstrating that non-erythropoietic EPO derivatives could also be tissue protective2 suggested the existence of a further non-haematopoietic receptor (EPOR)2, shared by members of the interleukin-3 receptor family.3 Nevertheless, studies in rodent models demonstrated that the expression of the classical EPOR was mandatory for EPO-induced neuroprotection.4
The hypothesis that EPO could specifically exert a protective effect on motor neurons arose from several lines of research. EPO was found to protect cultured motor neurons from serum-BDNF (brain-derived neurotrophic factor) deprivation or long-term kainate exposure.5 Treatment with EPO and non-erythropoietic derivatives improved motor performances and protected from motor neuron degeneration in the wobbler mouse model, although the effects were limited to women and survival was not prolonged.6 In SOD1G93A mice, increased EPO and EPOR expression in the brain cortex was interpreted as a possible compensatory effect of altered neuronal function.7 In the same model, EPO treatment delayed the onset of motor deterioration and protected thoracic spinal cord motor neurons from degeneration.8 However, this effect could not be replicated.9 EPO was also found to reduce SOD1 aggregates in motor neurons.10 In patients with amyotrophic lateral sclerosis (ALS), the cerebrospinal fluid EPO level was lower than in patients with other neurodegenerative diseases, and its concentration was suggested to correlate with the progression of the disease.11 Moreover, the cerebrospinal fluid concentration of EPO and vascular endothelial growth factor were found to be significantly higher and lower, respectively, in hypoxaemic patients with ALS, suggesting an intact common oxygen-sensor pathway.12 On the basis of the results of our previous pilot study,13 we designed a double-blind, randomised, placebo controlled trial to assess the efficacy of rhEPO as an add-on treatment in patients with sporadic ALS.
Methods
Patients
The trial was coordinated by the ALS Centre at the IRCCS Foundation ‘Carlo Besta’ of Milan. Consecutive patients were screened for eligibility at all the 25 Italian ALS centres. Patients aged 18–75 years and diagnosed with probable laboratory-supported, probable or definite ALS according to El Escorial revised criteria were eligible. Inclusion criteria were onset of weakness ≤18 months and slow vital capacity (sVC) ≥70% of predicted in seated position at screening visit. Exclusion criteria were haematocrit >51% in men and >49% in women, haemoglobin value >17 g/dL; non-invasive ventilation (NIV) >6 h daily; known familial ALS or first-degree relative with ALS; diagnosis of frontotemporal dementia; history and/or instrumental evidence of previous thrombotic vascular event or cardiac diseases; uncontrolled hypertension (systolic ≥160 mm Hg and diastolic ≥95 mm Hg irrespective of treatments at two consecutive evaluations); active solid or myeloproliferative malignancy; known hypercoagulable disorders. All patients were asked to continue riluzole 100 mg daily or to be on a stable dose for at least 30 days prior to screening. Patients who did not take riluzole at the screening visit were considered eligible and could be randomized; in this case, they could not start riluzole during the entire study period. The protocol was approved by the ethics committee at each site. All patients provided written informed consent before any study-related procedure.
Trial outcomes
The primary outcome was one single composite outcome of time from randomisation to death, tracheotomy or >23 h NIV daily for 14 consecutive days. Secondary outcomes were changed from baseline in the ALSFRS-R score, sVC in the seated position and patients’ quality of life measured by the ALSAQ-40 questionnaire. Tolerability was evaluated analysing adverse events (AEs) causing withdrawal.
Randomisation
Patients were randomly assigned to receive either intravenous rhEPO 40 000 IU or matching placebo (1:1 ratio) fortnightly for 12 months. Randomisation was stratified by centre, disease severity (ALSFRS-R ≤33 vs >33) and onset (spinal or bulbar), with a block size of four within each centre. The Neuroepidemiology Unit at the coordinating centre, independent of the study, generated the computer-based randomisation sequence known only to two staff persons and the drug dispenser. Treatment was allocated by a web-based randomisation system, available 24 h a day. The procedure incorporated eligibility checks according to protocol and was performed on request from the centres. The sequence was always available for emergency unmasking.
Masking
RhEPO (Eprex) was purchased from Janssen-Cilag SpA (Cologno Monzese, Italy) by the coordinating centre and directly shipped to the company (Pierrel Research IMP srl, Cantù, Italy) in charge of preparing the investigational drug (rhEPO or 1 cc of saline) in syringes of identical appearance, sealed in sequentially numbered identical containers according to the allocation sequence. Shipping was performed for each patient within 1 week after randomisation using a refrigerated express carrier. Each centre stored the investigational drug at −4°C.
Patients, neurologists, laboratory biologists/technicians (two at each centre) and a statistician were masked to treatment allocation and did not have access to any data related to rhEPO haematopoietic activity. Briefly, all participating centres generated a specific access code for the blood withdrawals of patients with ALS enrolled in the EPOS trial. At each treatment visit, the local laboratory unit received the de-identified vial reporting the randomisation number before treatment administration. Blood parameters were emailed to the coordinating unit where a trained person readily alerted the treating neurologist on the decision to administer or delay the treatment based on safety protocol parameters (see below). Patients and treating neurologists did not have access to any data on blood parameters, whereas laboratory personnel did not have access to any data allowing the identification of the patient. This procedure was elaborated to maintain the blindness of the study. Finally, when we designed the study, we reasoned that patients treated with rhEPO could have had a higher risk of treatment delay as defined by the safety protocol. Therefore, the randomisation centre generated a list of placebo patients randomly assigned to 1 week treatment delay balanced with the number of patients allocated on rhEPO treatment needing true delay at each centre. This procedure was elaborated to maintain the blindness of the study.
Procedures
After giving informed written consent, eligible patients underwent haematological examinations (haemochromocytometry, renal and liver function, serum iron, ferritin, transferrin, reticulocyte count, coagulation tests), blood pressure and body mass index (BMI) measurement and sVC, ALSFR-R and ALSQ40 assessment. Randomisation was performed within 15 days after the screening visit. At each fortnightly treatment visit, safety parameters (haematocrit >51% in men and >49% in women or haemoglobin value >17 g/dL and value raised >1 g/dL at the end of the interval between two subsequent doses), blood pressure and BMI were assessed. Symptoms of nocturnal hypoventilation (nocturnal arousals, morning headache, excessive daytime sleepiness, vivid dreams), medications and AE were actively monitored and recorded. ALSFR-R and sVC were assessed monthly. At the 6-month and study end or dropout visit, the patient also underwent complete haematological examinations and ALSAQ-40 assessment. At each treatment visit, treatment administration was allowed or delayed for 1 week by the trial coordinating office after assessment of safety parameters. If treatment was delayed, the following week safety parameters were repeated before drug administration. The delay of treatment administration for more than two times caused the dropout of the patient. After the study end at month 12, patients underwent monthly follow-up visits for a further 6 months to record primary outcome events. All centres were provided with a spirometer (Spirobank G multifunction, Medisan srl, Milano, Italy) and disposable tubes for sVC assessment, and trained in its use. All data were recorded by an electronic case record form specifically developed (Nubilaria srl, Novara, Italy). Trial monitoring was performed by an independent contract research organisation (CROM srl, Verona, Italy) that assured consistency in measuring outcomes across centres by scheduled site visits.
Co-treatments
Nutritional status and ventilation could affect survival and thus the primary outcome of the trial. During the first investigator meeting held in Milan on 6 June 2010, all participating centres agreed on the approach to cotreatments, sharing the opinion that the ultimate decision would be personal to each patient. We agreed that percutaneous endoscopic gastrostomy or an equivalent device should be proposed to all patients in the case of score 1 or 2 at item 3 of the ALSFRS-R, unintentional loss of body weight >10% in the past 3 months or choking during food, liquids or medication ingestion. NIV should be proposed to all patients in the case of score 0 or 1 at item 10 or 11 of the ALSFRS-R, sVC<50% or abnormal nocturnal oximetry (SaO2 <90% for 4% of the overnight recorded time).
Sample size estimation
On the basis of the results of our pilot study (ie, observed rates of 0.56 for death and 0.33 for tracheotomy at 18 months in the placebo group),13 we estimated that we would need a sample size of 203 patients followed up for 12 months to give 97% power to detect a significant difference between rhEPO and placebo corresponding to a 67% relative reduction of risk of death and 74% power to detect a 70% relative reduction of risk of respiratory events (tracheotomy or >23 h NIV), with a two-sided type 1 error of 5% and given an anticipated dropout rate of 10%.
Statistical analysis
The main analysis of primary and secondary outcomes included all randomised patients who took at least one dose of the investigational drug in their original assigned groups. All analyses were performed both for the entire population and for the subgroups of patients with ALSFRS-R ≥33 or <3314 and with spinal or bulbar onset at randomisation. A per-protocol analysis was carried out excluding non-compliers (patients who took <80% therapy). Demographic characteristics and clinical features of randomised patients at baseline were reported by treatment arm and compared using χ2 test, student t test, Wilcoxon rank-sum test or Fisher’s exact test as appropriate. Time from randomisation to death, tracheotomy or >23 h NIV daily for 14 consecutive days was analysed in terms of the annualised rate with the corresponding 95% CI, and p value using a χ2 test with one degree of freedom for rate comparison (based on Poisson regression). The Kaplan-Meier method was used to obtain survival curves with the corresponding log-rank test. A Cox model was applied to estimate the treatment effect in terms of HR with 95% CI, adjusted for sex, age, ALSFRS-R score at baseline and disease duration. The number of patients experiencing an AE causing withdrawal were reported and compared between the two groups by Kaplan-Meier curves of the time to withdrawal and the corresponding log-rank test. Change from baseline in sVC, ALSFRS-R and ALSAQ-40 was assayed by mixed effect models. All data were analysed using SAS V.9.3 (SAS Institute INC. Cary, North Carolina, USA). This study is registered with EudraCT, number 2009-016066-91 (EPOS trial).
Results
Between August 2010 and November 2012, 208 of 545 eligible patients (38%) were randomly allocated to the treatment arms (104 in the rhEPO arm and 104 in the placebo arm; figure 1). Patients were recruited at 25 Italian ALS centres from 13 regions (figure 2). Table 1 shows the demographic and clinical characteristics of the treatment groups at baseline. In the analysis of the primary outcome performed on all patients who received at least one dose of the interventional treatment (eg, 103 patients in the rhEPO arm and 97 patients in the placebo arm), the rate of events (death, tracheotomy, >23 h NIV) at 12-month follow-up did not differ between the treatment groups, even after stratification by disease severity and onset (table 2). The Kaplan-Meier analysis did not disclose any difference in terms of the log-rank test (p=0.99; figure 3) and Peto test (p=0.89). The corresponding HR between rhEPO and placebo, adjusted for gender, age, ALSFRS-R score and disease duration, was 1.02 (95% CI 0.57 to 1.83). The analysis of efficacy by intention-to-treat performed on all randomised patients (eg, 104 patients in the rhEPO and placebo groups) yielded the same results.

Flow chart of the EPOS trial. CONSORT flow diagram. Flow diagram showing the progress of patients throughout the EPOS trial. ALS, amyotrophic lateral sclerosis rhEPO; recombinant human erythropoietin.
Italian centres participating in the EPOS trial with a number of patients enrolled in brackets.
Demographic characteristics and clinical features of randomised patients at baseline in the two treatment groups
Analysis of efficacy for the primary outcome at 12-month follow-up

Primary outcome analysis. Survival probability in terms of death, tracheotomy and 23 h non-invasive ventilation for 14 consecutive days during the 12 months of the EPOS trial, with the corresponding p value of the log-rank test.
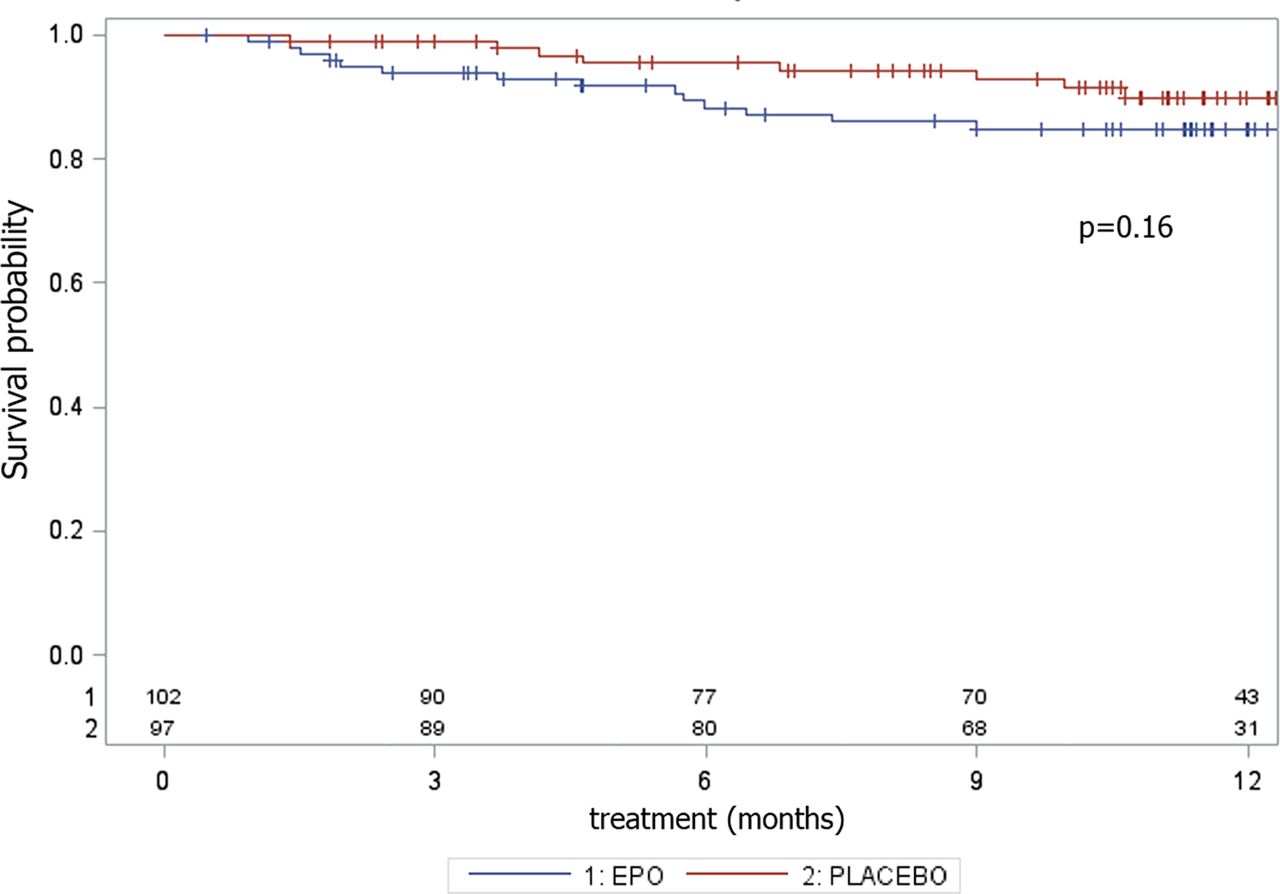
The percentage of AE causing withdrawal was twice as high in the rhEPO (16.5%) group as in the placebo (8.3%) group, although the difference was not significant in terms of the log-rank test (p=0.16), most likely due to the small number of events (table 3; figure 4). In the rhEPO group, we recorded four cases of deep venous thrombosis complicated in two cases by pulmonary embolism, two cases of cardiac arrhythmia, four cases of treatment suspension for >8 weeks and seven cases of delay more than two times due to altered safety parameters. In the placebo group, we recorded one case of deep venous thrombosis, one case of cardiac infarction, four cases of treatment suspension for >8 weeks and two cases of delay more than two times due to altered safety parameters (table 3).
Number and percentage of AEs causing withdrawal in the two treatment groups

Adverse event analysis. Survival probability in terms of adverse events causing withdrawal during the 12 months of the EPOS trial, with the corresponding p value of the log-rank test.
The analysis of the secondary outcomes during the 12 months of follow-up did not show differences between groups either in ALSFRS-R (p of mixed-effects models=0.31) and sVC (p of mixed-effects models=0.47) decline (figure 5) or ALSAQ-40 score (+29 points in the rhEPO group and +37 points in the placebo group from baseline to 12 months; p of mixed-effects models=0.23).

Secondary outcome analysis. Progression of ALSFRS-R (A) and slow vital capacity (sVC) (B) in the two treatment groups since the baseline visit through the 12-month trial period, with the corresponding p value of the repeated measure analyses.
All the above analyses were also performed for the subgroups of patients with ALSFRS-R ≥33 or <33 at randomisation and with spinal or bulbar onset, and per-protocol, and did not show any significant difference between the two treatment groups (data not shown). Haemoglobin and haematocrit values overlapped in the rhEPO and placebo groups at baseline, whereas they were significantly (p<0.01) higher in the rhEPO group than the placebo group throughout the entire study period (figure 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Haematological effects of recombinant human erythropoietin (rhEPO) mean values of haemoglobin (A) and hematocrit (B) in the two treatment groups since baseline visit through the 12-month trial period. At baseline, haemoglobin and haematocrit values overlapped, whereas they were significantly higher (p<0.01) in the rhEPO group than in the placebo group at 3, 6, 9 and 12-month follow-up.
At 18-month follow-up, 6 months after the treatment was stopped, 41 (41%; 3 lost to follow-up) of 100 patients in the rhEPO group and 31 (33.3%; 4 lost to follow-up) of 93 placebo patients reached the primary outcome (death or tracheotomy).
Discussion
Several studies suggested that EPO can promote the homeostasis of cells under stress and exert protective actions on different tissues. Peripheral administration allows EPO to penetrate through an intact blood-brain-barrier,15–17 and this has been exploited to test its neurotrophic effects in multiple sclerosis, Parkinson's disease and schizophrenia.18 ,19 Encouraging preclinical studies with EPO and its non-erythropoietic derivatives in models of central and peripheral nervous system degenerative diseases,2 expression of the non-erythropoietic receptor (EPOR)2 in motor neurons6 and preliminary data from ALS mouse models8 and a pilot clinical trial13 suggested that patients with ALS might benefit from rhEPO treatment. Conversely, our study demonstrated that rhEPO administered at the dose of 40 000 IU fortnightly did not change either survival or disability at 12 months. Patients’ demographic and clinical features were well balanced between the treatment arms, supporting the reliability of the results. The significant and stable increase of haemoglobin and haematocrit values in the rhEPO group throughout the entire treatment period demonstrated that rhEPO exerted its expected haematological effects, and therefore it was not degraded in patients with ALS. This observation strengthens our negative findings, suggesting that the lack of neuroprotective effect could not be attributed to an altered biological activity of rhEPO at the haematological level.
Our negative findings appear to be in keeping with those disappointing from previous clinical studies investigating rhEPO neuroprotection in critical illness and patients with stroke.1 RhEPO may remain a promising treatment in schizophrenia19 and in the prevention of cognitive impairment after cardiac surgery and cardiopulmonary bypass,20 though these preliminary data need larger confirmatory studies. Therefore, despite the bulk of preclinical findings in favour of a substantial protective activity of EPO outside the bone marrow, no evidence is currently available to support the hypothesis that rhEPO can rescue injured neurons in patients with acute or chronic progressive neurological diseases like stroke and ALS.
In most previous clinical studies of neuroprotection, cardioprotection and renal protection, rhEPO was acutely administered at doses ranging from 40 000 IU daily for 3 days to 50 000 IU 24 and 48 h after the event, whereas in critically ill patients the schedule was 40 000 IU weekly for 3 weeks.1 Ours is the first trial in which rhEPO was chronically administered to non-anaemic patients with a degenerative disease for a 12-month period. One limitation may appear to be the lack of a previous dose-finding study. However, we have chosen the dosing schedule of rhEPO 40 000 IU fortnightly based on the pharmacokinetic profile, the known linear relationship between a single dose administered and erythropoietic response, and the turnover of reticulocytes,21 with the aim of reducing the thrombotic risk in patients. Moreover, acute higher doses used in previous clinical studies increased the rate of thrombotic vascular events.1 40 000 IU approximately equals one-third of the maximally effective single dose in a 70 kg participant (eg, 1800 IU/kg). In our study, it did not significantly increase the rate of thrombotic complications compared with the placebo group, most likely due to the small number of events, being the overall number of AEs twice as high as in the rhEPO treated group. In rodent models,22 the dose of 2500 IU/kg/day was reported to be the most effective for neuroprotection but caused an increase of haematocrit value that would not be acceptable in humans.
A recent reappraisal of patients’ clinical features included in ALS trials suggested that the intrinsic limitations imposed by a classical randomised clinical trial can lead to the exclusion of patients representing the ALS population in clinical practice, reducing the reliability of the results.23 It has been suggested that the enrolment of patients in the earliest phases of ALS could increase the probability of identifying successful disease-modifying treatments. In our study, the percentage of patients excluded due to respiratory insufficiency (15%) and of those who did not reach the diagnostic certainty level of probable ALS according to the revised El Escorial criteria (1.5%) at randomisation was small compared with previous trials. Similarly, gender distribution was well balanced between arms, thus avoiding the underrepresentation of women observed in other trials.23 However, the mean age of ALS onset was slightly lower than that recorded in epidemiological studies,24–27 possibly accounting for the lower 1-year death rates.
Like previous trials in ALS, results from our pilot study did not replicate in the larger phase III trial. In particular, in the pilot study, we had observed a higher prevalence of primary outcome events (death and tracheotomy) in the placebo group at 18 months follow-up, most likely a chance result due to the small sample size. In ALS, the most important goal of new treatments which can protect motor neurons and axons from progressive degeneration in a time frame that is useful to patients remains far from be achievement. Our phase III randomised trial demonstrated that rhEPO does not have any positive effect on the course of ALS, lengthening the list of disappointing results from all the previous studies.28–37
Acknowledgments
The authors thank all the patients participating in the trial and their families. The authors thank all the collaborating nurses. The authors thank Laura Mastrosimone, Domenico Arenella and Laura Ferradini of the EPOS trial coordinating centre and Ilaria Riela for administrative and legal assistance. The authors also acknowledge the Fondazione Italiana di per la SLA—Sclerosi Laterale Amiotrofica.
References
Footnotes
Collaborators EPOS trial study group: Daniele Cazzato (‘Carlo Besta’ Neurological Institute, Milan); Edward Cesnik, Elisabetta Groppo, Elisabetta Sette (Ferrara); Carla Pani, Emanuela Costantino, Francesco Orlandini, Daniela Boi (Cagliari); Giorgia Querin, Carla D'Ascenzo (Padua); Anna Sagnelli, Giovanni Piccirillo (Naples); Marina Aiello, Alfredo Chetta, Andrea Grassi (Parma); Christian Lunetta, Eleonora Maestri (NEMO Centre, Milan); Alessandro Padovani, Mariasofia Cotelli, Alice Todeschini (Brescia); Stefania Morino, Antonella Di Pasquale, Pamela Latino (Rome); Stefania Casali, Stefania Battistini, Marilena Pirrelli (Siena); Roberto Cantello, Nicola Nasuelli, Serena Servo (Novara); Riccardo De Gennaro, Ernesto Gastaldo (Mestre); Eleni Georgoulopoulou, Nicola Fini (Modena); Alfonsa C. Taiello, Tiziana Colletti (Palermo); Andrea Calvo, Cristina Moglia, Giuseppe Fuda (Torino); Kalliopi Marinou (‘S.Maugeri’, Milan); Nilo Riva, Federica Cerri, Ignazio D. Lopez (S. Raffaele Hospital, Milan); Domenico De Cicco, Gianluca Battaglia (‘S.Maugeri’, Mistretta); Norina Marcello, Manuela Rinaldi (Reggio Emilia); Carlo Scialò, Vittorio Mantero, Maria Mascolo (Genova); Cecilia Carlesi, Elena Caldarazzo Ienco (Pisa); Antonio Di Muzio (Chieti); Lorenzo Verriello, Delia D'Amico (Udine); Isabella L. Simone, Rosanna Tortelli, Rosa Cortese (Bari), Ilaria Bartolomei (Bologna).
Contributors GL and GF have designed the study; GL was the principal investigator and wrote the manuscript; GF was responsible for the randomisation unit; IT has performed the statistical analyses; GA, GB, MC, CC, AC, MC, RE, RF, MF, FG, EG, VLB, GL, JM, LM, MRM, GM, VP, RQ, RR, FS, GS, GS, PV acted as site principal investigators, participated at the investigator meetings and definition of the final protocol, and approved the final manuscript; EDB and all the neurologists listed in the EPOS trial study group have actively participated in the enrolment of patients and assessment of the outcomes, and have read and approved the final manuscript.
Funding The study was funded by the IRCCS Foundation ‘Carlo Besta’ Neurological Institute, Milan, Italy, an institution of research and care of public body. The study database is held by the Neuroepidemiology Unit.
Competing interests None.
Patient consent Obtained.
Ethics approval Each of the 25 participating centres obtained approval from the local ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.