Article Text

Abstract
Objective With the emergence of experimental therapies for Duchenne muscular dystrophy (DMD), it is fundamental to understand the natural history of this disorder to properly design clinical trials. The aims of this study were to assess the effects produced on motor function by different DMD genotypes and early initiation of glucocorticoids.
Methods Through the NorthStar Network, standardised clinical data including the NorthStar Ambulatory Assessment score (NSAA) on 513 ambulant UK boys with DMD were analysed from 2004 to 2012. For the analysis of the genetic subpopulation, we also included data from 172 Italian boys with DMD. NSAA raw scores were converted into linear scores.
Results On the linearised NSAA, we observed an average decline of 8 units/year (4 units on raw NSAA analysis) after age 7. The median age at loss of ambulation (LOA) was 13 years (95% CI 12.1 to 13.5); 2 years prior to LOA, the estimated mean linearised NSAA score was 42/100 (13/34 raw scale). Starting glucocorticoids between 3 and 5 years conferred an additional gain in motor function of 3 units/year (1.3 raw units) up to age 7. When analysing the effect of genotype in the UK and Italian cumulative cohorts, individuals with deletions amenable to exons 44 and 46 skipping declined at a slower rate over 2 years (9 units (4 raw units), p<0.001), while 53 and 51 skippable deletions showed a faster decline of 14 (4.5; p<0.001) and 5 linearised units (2.4 NSAA units; p=0.02), respectively.
Conclusions Our study provides a novel insight on the current natural history of DMD, which will be instrumental for the design of future clinical trials.
- NEUROMUSCULAR
- MUSCULAR DYSTROPHY
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Duchenne muscular dystrophy (DMD) is a life-limiting neuromuscular disorder, affecting 1/5000 live males,1 which results in progressive weakness, loss of ambulation (LOA) and death by the third–fourth decade of life.2 ,3 In recent years, a number of experimental therapeutic approaches have been developed aiming at restoring the absent dystrophin protein in muscles, including: exon skipping by antisense oligomers,4 ,5 suppression of stop codon mutations,6 ,7 and gene therapy8 ,9 targeting specific mutations of the dystrophin gene. Other non-mutation-dependent approaches include idebenone, a potent antioxidant;10 tadalafil, which affects muscle blood flow; and the upregulation of utrophin, a molecule which may compensate for the lack of dystrophin in skeletal muscle.11
The overall natural history of DMD has changed significantly in recent years. With the introduction of glucocorticoid (GC) therapy and systematic implementation of the standards of care, LOA and life expectancy are gradually shifting to a later age, requiring continuous monitoring of the evolving clinical course DMD.12–15 In addition, mutation-specific therapeutic approaches have generated the need for understanding the natural progression of the targeted genotype subgroups, especially if differences from the overall DMD population exist. It is recognised, for example, that some patients with mutations skippable by exon 44 or 45 may present an intermediate phenotype, due to an elevated number of revertant fibres and residual dystrophin expression;16 ,17 however, the influence of the different genotypes on disease course appears to be modest according to a recent study.18 Furthermore, the course of DMD can also be influenced by age at the starting GC and GC regimen.19 ,20 The standards of care suggest initiating GC in the plateau phase;2 ,3 the additional benefit of starting therapy at a younger age has been reported in a small case-series of children21 which invites further exploration.
Clinical trials design of rare progressive disorders, such as DMD, poses various challenges: (1) the number of patients who can be recruited into studies is often limited. In case of exon skipping, recruitment is mutation-specific, making it problematic to run large and well-powered studies; (2) cohorts may be too small to allow for a placebo arm, and small cohorts may not be representative of the natural variability even within a genotype, thereby leading to skewed results; (3) validated outcome measures may only capture a snapshot of the progression of the disorder and not be applicable to different stages of the condition, further limiting the number of patients recruitable into studies (eg, the 6 min walk distance test (MWD),22 a validated primary outcome measure in a number of clinical trials, can only be performed in ambulant individuals); (4) most of the future genetic therapy approaches will target small cohorts due to the high costs of experimental therapies and challenges of running large multinational studies and will also need long-term follow-up to allow ascertainment of potential side effects, thereby emphasising the need for precise subgroup characterisation.
Large natural history studies, if systematically conducted, can meaningfully inform the design of clinical trials, by offering an up-to-date description of the progression of the condition according to concurrent standards of care, and a valid platform of natural history data to guide both inclusion criteria and outcome measure selection.
In our study, which focused on the NorthStar Ambulatory Assessment (NSAA) as a measure of motor function, our objectives were as follows: (1) to assess the NSAA evolution in ambulant boys with DMD in the UK treated according to the agreed standards of care; (2) to describe the rate of progression of a subgroup of young boys with DMD in the UK treated with GC below 5 years of age; (3) to describe the NSAA rate of decline in DMD stratified for DMD genetic mutations including individuals in the UK and in the Italian neuromuscular clinical network.
Methods
Through the UK NorthStar Network and database, which encompasses 17 neuromuscular centres, clinical data from 2004 to 2012 on 513 ambulant boys with DMD between 3 and 16 years of age (mean age 9.5 years) were included in the analysis. The database systematically collects clinical information on boys with DMD in the UK. The diagnosis is confirmed in most cases by DNA diagnostic technique covering all DMD gene exons and/or a muscle biopsy. Mutations were classified according to the Leiden Muscular Dystrophy database.23 All patients included in the analysis were treated according to the standards of care,2 ,3 comprising therapy with GC administered either as daily prednisolone/deflazacort or intermittently (ie, prednisolone 10 days on:10 days off or alternate days19 ,20).
For the analysis of the young patient with DMD, we addressed the evolution in UK boys from 3 years onwards, comparing those who started GC before and after 5 years of age, as this would be a relevant population for future clinical trials.
NorthStar Ambulatory Assessment
The NSAA is a validated unidimensional functional scale for ambulant boys with DMD.24 ,25 The scale is suitable for multicentric studies,24–26 and is widely used internationally, in clinical settings and as secondary outcome measures in clinical trials. Both traditional and modern psychometric (Rasch) analysis has confirmed this to be a robust scale.27 Recently, an updated linearised version of the scale has been developed following an in-depth Rasch analysis, to improve the interpretation and capturing of clinically meaningful changes across the breadth of the scale.28
Statistical methods for the UK data set
We used frequencies and percentages to describe the number of boys with each mutation. We described the median and other percentiles for age at LOA, along with 95% CI using the Kaplan-Meier method.
Previous work showed a difference in the relationship between motor function and age, up to 7 years of age compared with later on in childhood;13–15 ,18 ,20 ,29 ,30 therefore, we decided to look at these two time periods separately for the purposes of this study.
NSAA score was our primary outcome; additionally, we converted the raw NSAA total scores into linearised scores. Using the Rasch methodology reported recently,28 the conversion was performed via a logit transformation, and this formed a linearised score on a 0–100 scale. We used this linearised transformation of the NSAA scale in all subsequent analyses and present the results in terms of this linearisation. The benefit of this approach is that the change score means the same across the breadth of the scale. Furthermore, analyses were repeated for the raw NSAA score scale and results are presented in brackets as raw scores are perhaps more readily understood by clinicians. The tranformed score was calculated using the total NSAA score minus 2 (ie. the score for “lifts head” as this item misfits the construct for ambulation) Now out of 32, the raw scores were converted to 100 points on the new scale as reported.28 For some children, the lifts head component of the NSAA scale was missing; therefore, it was not possible to convert their total NSSA score to the linearised score. We performed a sensitivity analysis considering several possible values for the missing lifts head score and derived the corresponding linearised scale. We found that the results from these analyses were robust and there was no impact on subsequent findings. We fitted a separate multilevel model for each age period (data over age 7 years and data under 7 years), with a random effect for patient, nested within a random effect for centre. We modelled the relationship between NSAA and age and adjusted for age at start of GC and treatment regimen. In addition, by including interaction terms in our models, we explored whether the rate of deterioration after age 7 or improvement in motor function up to age 7 varied according to the type of mutation: duplication, deletion or point mutation. For the early time period, we explored whether there was a difference in improvement in NSAA between those boys who started treatment early (before age 5) and those who started later (between 5 and 6.5 years), by fitting an interaction term. Simple comparisons for items in the NSAA scale were made between early and late starters using t tests. Similarly, BMI z scores31 were compared between early and late starters.
Fractional polynomials were used to check for any non-linearity in the relationship between independent factors and NSAA outcome.
Populations with skippable deletions—UK and Italian data sets
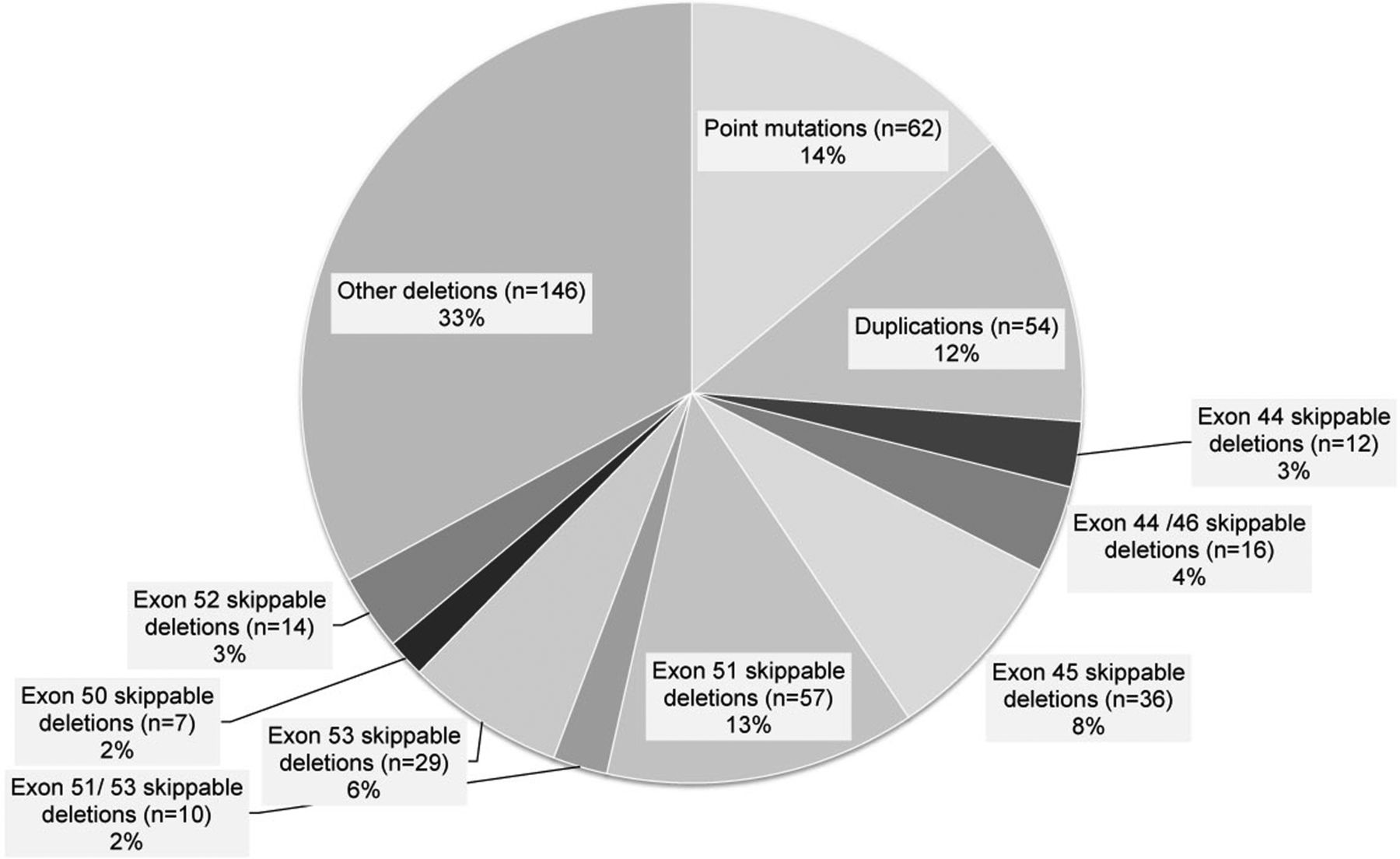
Genetic information was available for 442 of 513 UK boys (figure 1). Mimicking the setting of phase II and phase III clinical trials for the subanalysis of the skippable genotypes, we included:

Genotype breakdown of individuals registered in the UK NorthStar Clinical database. Genetic information was available for 442 UK boys registered in the NorthStar database.
(1) All boys with DMD >5 years of age; (2) with a transformed NSAA total score of 52/100 at time 0/baseline (corresponding to ∼230 m 6MWD;12 (3) and on stable GC for a minimum of 3 months at baseline; (4) boys with at least 24 months of longitudinal data.
This resulted in a reduced data set of 223 boys with DMD from UK, which was combined with 172 boys with DMD with the follow-up by the neuromuscular Italian clinical network coordinated by the Department of Paediatric Neurology, Catholic University, Rome. Of the Italian boys with DMD, 74 had specific skippable deletions: 20 with deletion exons 44/46 skippable; 1 with exon 44 skippable; 17 with exon 51 skippable, 1 with exon 51 or 53 skippable; 25 with exon 53 skippable; 8 with exon 50 skippable; 2 with exon 52 skippable.
Statistical methods for skippable populations
We compared the change in linearised NSAA over time between skippable exons for the UK and Italian data combined. For the Italian patients, data were available at fixed time points, at 12 months and 24 months follow-up. We created a similar format of data from the UK NorthStar database using fixed data±1 month and combined the two data sets together (n=223+172=395 DMD). Since a small minority of boys with DMD can benefit from skipping more than one exon, we fitted a series of multilevel models, each including two interaction terms to compare the rate of decline in NSAA between those boys with and without a particular exon skip. The first interaction term compared the decline over 12 months, and the second the decline over 24 months. All models included a random effect for patients nested within the country; we adjusted for age at the follow-up and treatment regimen.
All analyses were conducted in Stata, and for all tests a p value of <0·05 was considered statistically significant.32
Results
UK NorthStar data set
Age at LOA
In the UK NorthStar database, ambulation was lost in 137/513 boys between 9.5 and 16 years, including all GC treatment regimens. The median LOA was 13 years (95% CI 12.1 to 13.5). We also calculated the 10th centile: 9.5 years (95% CI 9.1 to 9.9); the 25th centile: 10.9 years (95% CI 10.1 to 11.1); and the 75th centile: 16 years (95% CI 15 to na).
NSAA slope of decline in boys >7 years of age
It has been previously reported that DMD boys gain motor function up to 7 years of age, after which they start to decline.13 ,14 ,20 ,22 ,29 ,30 In our cohort, the overall slope coefficient was −7.8 (95% CI −8.8 to −6.9; −3.7, 95% CI −4.1 to −3.3), meaning that our DMD population, on average, lost 8 linearised NSAA units/year after age 7 (figure 2). The estimated linearised NSAA score at age 7 was 73.4 (95% CI 70.3 to 76.5; 27.4, 95% CI 26.1 to 28.6). When compared to the whole DMD population, a positive interaction coefficient 0.7 (95% CI −1.9 to 3.3; raw NSAA:0.5 (95% CI −0.7 to 1.7)) suggested a possible trend for a slower decline in boys with duplications (p=0.5), and a trend towards a more rapid decline in boys with point mutations (interaction coefficient=−1.5; 95% CI −4.1 to 1.1; p=0.2; raw NSAA:−0.3 95% CI −1.5 to 1.0); however, neither of these differences was significant.

Linearised NorthStar Ambulatory Assessment (NSAA) slope of decline in boys > 7 years of age. With an average linearised NSAA score of 73.4 (95% CI 70.3 to 76.5) at age 7 (raw NSAA: 27.4, 95% CI 26.1 to 28.6), the overall slope coefficient was −7.8 (95% CI −8.8 to −6.9; raw NSAA: −3.7, 95% CI −4.1 to −3.3), meaning that our Duchenne muscular dystrophy (DMD) population on average lost 8 linearised NSAA units for each year, after age 7. At 8 years of age, the mean linearised NSAA was 65.5 (95% CI 62.7 to 68.4; 23.7, 95% CI 22.6 to 24.8), while at 10 years of age it was estimated to be about 49.7 units (95% CI 46.8 to 52.8; 16.3, 95% CI 15.1 to 17.5). The mean linearised NSAA score was 42 units and 34 units 24 and 12 months before losing ambulation (equivalent to 13 and 9 raw scores, respectively).
In order to determine which NSAA score may best predict LOA within 24 and 12 months, assuming a mean LOA of 13 years, we estimated the mean linearised NSAA score from our model, which in our population was 41.9 units (95% CI 38.5 to 45.3; 12.6, 95% CI 11.2 to 14.1) 2 years prior to losing ambulation, and 34 units (95% CI 30.1 to 38.0) (8.9, 95% CI 7.2 to 10.7) 1 year prior to LOA.
NSAA in young boys with DMD
Prior to 7 years of age, young DMD boys improve their motor function, with an overall gain of 4.0 linearised units/year (95% CI 2.2 to 5.8 p<0.001; 1.4, 95% CI 0.6 to 2.1), adjusting for the GC treatment regimen. As part of this analysis, we explored the impact that starting GC before age 5 may have on DMD as measured by the NSAA. When comparing 78 DMD boys, who started daily/intermittent GC before age 5 (mean age at start=4.5 years) with 163 boys who started GC between ages 5 and 6.5 (mean age at start=5.7 years), the coefficient of interaction was −2.7, (95% CI −6.8 to 1.3, p=0.2), favouring early starters by almost 3 linearised units a year (figure 3; Raw NSAA:−1.3, 95% CI −3.0 to 0.3, p=0.1). By age 7, the mean total NSAA was different between the two groups (p<0.01): 73.8 (95% CI 67.5 to 80.1; 27.0, 95% CI 24.6 to 29.4) in early starters and 68.7 (95% CI 64.1 to 73.4; 25.1, 95% CI 23.2 to 26.9) in the late-starter group.
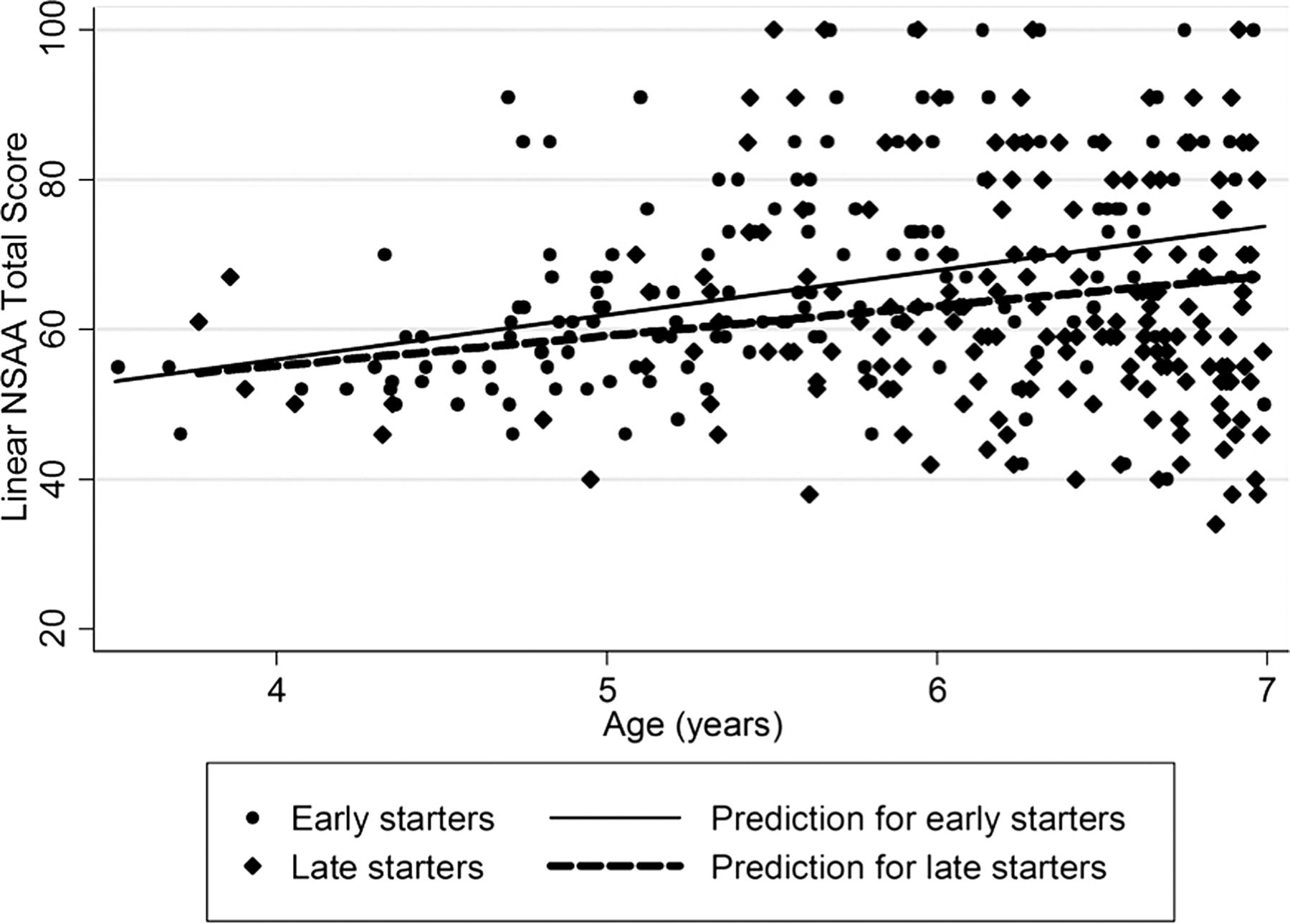
NorthStar Ambulatory Assessment (NSAA) in boys with Duchenne muscular dystrophy (DMD) <7 years of age. The interaction coefficient between 78 boys with DMD who started daily or intermittent glucocorticoid (GC) before the age of 5 with 163 boys who started steroids between ages 5 and 6.5 years was −2.7 (95% CI −6.8 to 1.3, p=0.2; raw NSAA: −1.3, 95% CI −3.0 to 0.3), p=0.1 favouring early starters. By age 7, the mean total NSAA was 73.8 (95% CI 67.5 to 80.1) in early starters and 68.7 (95% CI 64.1 to 73.4) in late starters (p<0.01; raw NSAA: (27.0, 95% CI 24.6 to 29.4 in early starters and 25.1, 95% CI 23.2 to 26.9 in late starters).
A difference between the two groups was observed in the following items of the scale: standing on heels (p=0.002), jumping (p=0.004), hopping (p=0.001), lifting head (p=0.009), standing to sit (p=0.008) and the 10 m run (p=0.05). The mean SD BMI for early starters and late starters showed no statistical difference at 7 years (p=0.2).
NSAA in UK and Italian skippable populations
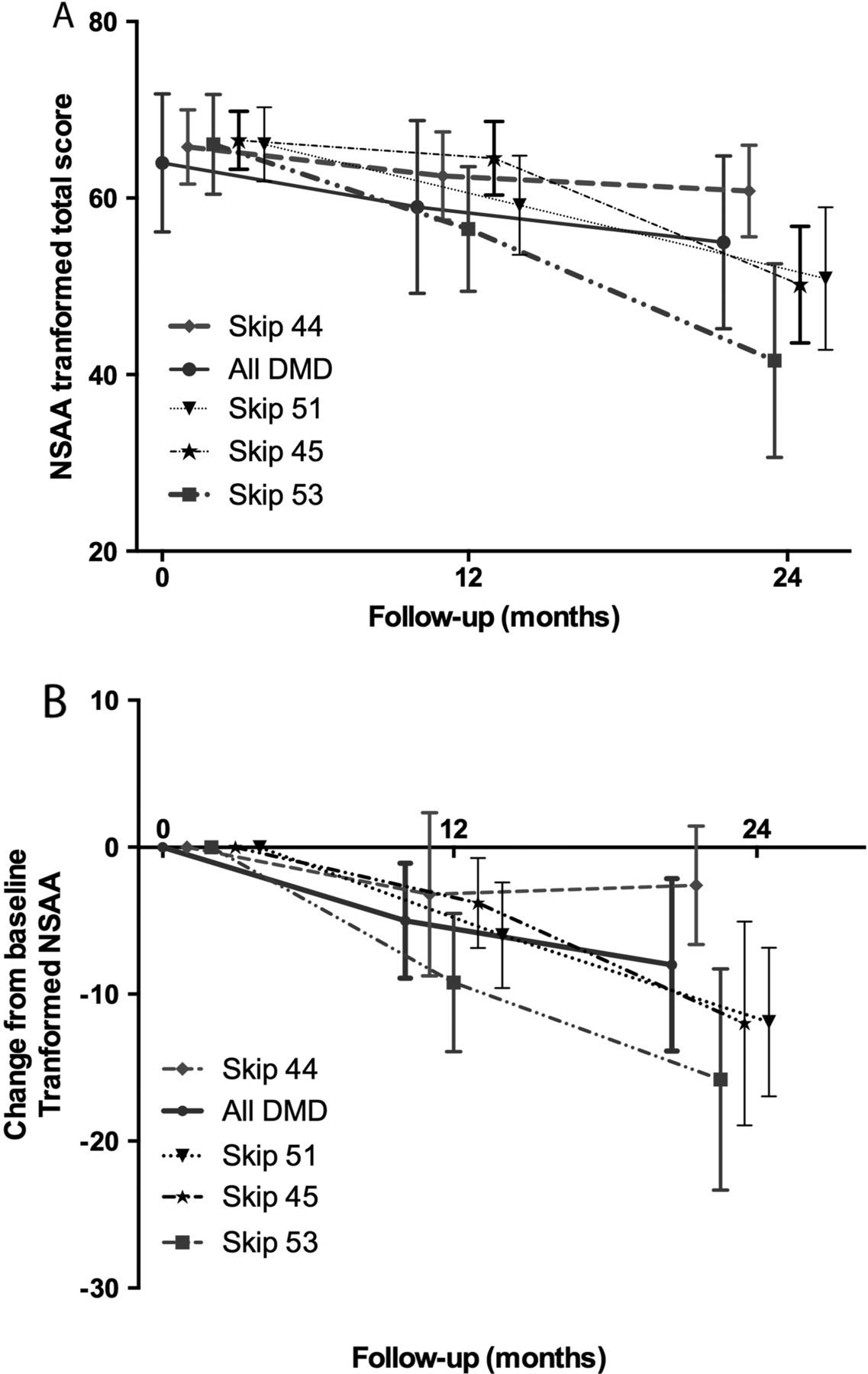
For this subanalysis, we included 395 DMD (ie, 223 UK+172 Italian) boys meeting the age/motor function and GC requirement for inclusion criteria in most of the ongoing clinical trials as described in the methods above. The whole population showed a decline of 5 units (95% CI−6.5 to −3.5; p<0.001) at 12 months and 13.8 (95% CI −15.7 to −12.0; p<0.001) at 24 months. When compared to the whole DMD population, boys skippable by exons 44 (n=27) and 46 (n=34) showed a slower decline, which became significant at 24 months follow-up (table 1 and figure 4). The interaction coefficient for the population skippable by exon 44 was 2.5 (95% CI −3.2 to 8.3) at 1 year (p=0.39) and 9.1 (95% CI 2.3 to 15.9) at 24 months (p<0.001), suggesting that over the course of 2 years boys with exon 44 skippable deletions lost nine points less on the transformed NSAA scale than the remaining DMD population (table 1A and figure 4). Similarly, boys skippable by exon 46 showed a slower decline at 24 months with an interaction coefficient of 8.8 (95% CI 2.6 to 15.1; p<0.01). On the other hand, the negative interaction coefficient for the population skippable by exon 53 (n=41) was suggestive of a more rapid decline overall: −6.5 (95% CI −11.2 to −1.7; p<0.01) at 12 months and−14.2 (95% CI −19.9 to −8.5; p<0.001) at 24 months (table 1A and figure 4). Similarly, boys skippable by exon 51 (n=61) showed an increased loss of 5.7 linearised NSAA units at 2 years (interaction coefficient −5.7, p=0.02). We report the raw NSAA scores and interaction coefficients in table 1B. All the above comparisons are adjusted for treatment and age at follow-up. We found no significant difference among the remaining populations.
Linearised (A) and raw (B) NSAA total score: interactions coefficients for skippable genotypes at 12 and 24 months
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean (95% CI) linearised NorthStar Ambulatory Assessment (NSAA) total score mean (A) and change from baseline (B) for skippable genotypes at 12 and 24 months. The overall slope of decline over 24 months is described in boys with Duchenne muscular dystrophy (DMD) >5 years of age, with a minimum NSAA of 52/100 (raw NSAA: 17/34), and at least 3 months on glucocorticoids at baseline. (For illustration purposes only, the plots are shifted to avoid overlap of the 95% CI bars).
Discussion
In this study, we address a number of important issues regarding the progression of DMD, which inform patients/families and the design and interpretation of experimental therapies.
We recently reported on the general characteristics of children followed in the UK Northstar clinical network.20 In the current study, we describe the effect of age on disease progression on a linearised NSAA scale; we explore the effect of initiating GC at a younger age than the current guidelines; in addition, we assess the possible influence of DMD genotype on disease progression by gathering data from a very large group of UK and Italian boys with DMD stratified by genomic mutations.
The benefit of the linearised scores is that the change score means the same across the breadth of the scale, whereas with the raw score a drop of several points at a mid-level of ability might actually mean a small loss of function (difficulty getting on and off a small step but still independently) and a drop of one point at either end of the scale might suddenly mean a loss of independence rising from the floor or loss of ability to run.
To characterise the disease on linearised NSAA, through the collaboration of the UK NorthStar clinical network, we report the course of DMD in >500 GC-treated UK boys. We describe the slope of progression after age 7, when motor function starts to decline. With an average linearised NSAA score of 73 at age 7 (=27/34 in the raw scale), we observed that the overall rate of decline on the transformed NSAA scale is 8 linearised units/year. Recent data suggest that in the linearised NSAA scale, ∼10 units are considered to capture a significant clinically meaningful change, irrespective of the scores along the scale that drive such a loss.28 This loss can mean the loss of the ability to stand on one leg or get up from the floor independently. Therefore, the linearised NSAA can reliably support clinical trials as a secondary outcome measure for ambulant DMD, with the potential to capture either stabilisation from the predicted 8-unit loss, or potentially even an improvement from baseline during the course of a 1-year study. When conducting clinical trials for ambulant boys, one possible outcome is that the boys may lose ambulation during the course of the study, affecting the overall interpretation of the study. From our cohort of NorthStar-registered individuals, we found that in the 137 UK boys who lost ambulation between 9.5 and 16 years, the median age was 13 years (95% CI 12.1 to 13.5). However, as age alone cannot serve as a predictor of LOA, we also report the estimated mean linearised NSAA score at 12 and 24 months prior to LOA (ie, at ages 12 and 11 years, respectively), which in our database were 34 and 42 units (approximately 9 and 13 in the raw scale, respectively). When selecting inclusion criteria for clinical trials in ambulant boys, a baseline total linearised NSAA score of 50 (∼16 of the raw scale) would therefore give reassurance that the boys are very unlikely to lose ambulation over the course of a 2-year study. Mazzone et al12 reported that in their Italian cohort followed for 24 months, the equivalent assurance was given by performing 330 m in the 6MWD. Only 2% of the boys, who at baseline walked at least 330 m, lost ambulation in the subsequent 2 years. Equally in their cohort of boys, approximately 98% of boys with an NSAA score of 17/34 (∼52 units in the linearised NSAA) were ambulant after 2 years, confirming our observation.12
If recruiting older patients with DMD in clinical trials poses the problem that they may lose ambulation during the course of the study, recruiting younger individuals poses the opposite problem: a number of patients with DMD improve their motor function up to age 7,13 ,15 ,20 ,29 thereby complicating the interpretation of a potential drug effect against the natural disease course. In our study, we report a functional gain of ∼3 units/year between 4–7 years (∼1.5 raw NSAA), with a mean NSAA score at age 7 of 73 (p=0.02; =27/34). We further report an improvement of 3 additional linearised units/year in 36 boys who had started GC <5 years (mean age: 3.4 years) when compared with boys who started GC >5, with a difference of 10 units by age 7 (p<0.01) but no difference in SD BMI in this early phase of therapy. We lack sufficient longitudinal data to conclude that this initial benefit is also sustained during the decline phase and that the age of LOA is further postponed, although data from smaller cohorts also appear to substantiate an increased efficacy of early GC initiation.21 Additionally, we lack sufficient longitudinal data to show if the cumulative GC therapy has an enhanced detrimental effect on the side effect profile. However, despite the limitation of our relatively small cohort (n=78), the positive impact of early initiation of GC therapy is of interest and demands future exploration. Furthermore, our analysis provides an insight on the motor function in this age group of patients, who are likely candidates for future clinical trials.
As part of our study, we also explore progression of disease in mutations amenable to genetic therapies. A number of current experimental approaches for DMD are mutation specific,4 ,8 ,9 ,33–35 emphasising the importance of investigating, within the disease spectrum, possible existing variations in the natural course of each genetic subtype, which could impact on clinical trials. Exon skipping by antisense oligomer is now in phase III studies for exon 51; molecules targeting exons 44, 45 and 53 are undergoing early clinical studies. In our study, we observed that individuals with duplications deteriorate minimally slower (∼1 unit/year) when compared with individuals with point mutations and deletions; however, this did not meet significance. In a substudy, we combined the Italian and UK data sets, to focus the analysis on skippable deletions and reproduce the setting of a clinical trial. We observed that boys skippable by exon 53 do progress more rapidly, losing an additional 14 units over 2 years (∼4–5 raw units), when compared with the whole DMD population (p=0.001). Similarly, individuals skippable by exon 51 declined faster, losing an additional 5 units (2.5 raw units; p=0.02). In contrast, boys with deletions skippable by exon 44 showed a less rapid decline measured as nine linearised units over 24 months (p<0.001; ∼4 raw units). Within the spectrum of severity of DMD, variability in phenotype among skippable deletions has been previously reported. Pane et al36 observed that boys skippable by exon 44 had better baseline results in the 6MWD and less drastic changes over the course of 1 year when compared with boys skippable by exon 53. Despite a similar trend, their results at 1 year did not reach statistical significance. The results of this study are consistent with this observation, as we could not detect a significant difference at 12 months, but only at 24 months. Similarly, Servais et al37 reported that patients treatable by exon 53 skipping have a more severe phenotype when upper limb function was assessed in a cohort of 14 DMD; additionally, LOA had occurred 1 year earlier in 90 boys, 53 skippable, when compared with 400 other boys with DMD. Residual dystrophin level can partially explain such an observed phenomenon. Recent studies showed that a larger presence of revertant fibres, which may favour a better outcome, is found in patients with mutations in the region of exons 45–47,16 ,17 when compared with levels of dystrophin found in patients with mutations in the region of exons 47–55. Residual dystrophin levels can therefore play a role as a weak modifier of DMD progression, although further exploration in larger cohorts should be performed in order to power studies precisely. The size effect of these differences is small and in practice negligible for studies lasting 1 year or less, but should be considered for those of longer duration. This might be particularly important in clinical trials where a treatment arm is compared to natural history data or a subset of patients. In particular, our data would discourage the recruitment of patients with deletions skippable for exon 44 as a comparator group for patients with 53 skippable deletions, and vice versa. Moreover, other disease modifiers are increasingly being reported in DMD, including polymorphisms in SPP138 ,39 and LTBP4.40 This latter gene was very recently independently validated in separate patient with DMD populations.41 When interpreting response to treatment, the combination of disease modifiers should therefore be accounted for.
In summary, our study describes up-to-date natural history data of the linearised NSAA scale in a large cohort of boys with DMD in the UK and Italy. The NSAA is currently included in clinical trials as a secondary outcome measure. Increased knowledge of the natural course of the disorder and its covariates (ie, age at starting GC and gene mutations), which can influence the sensitivity of clinical trials, will help in the design of future studies and interpretation of their results.
Acknowledgments
The support of the Muscular Dystrophy Campaign to the North Star Network, and of the MRC Neuromuscular Translational Research grant is gratefully acknowledged. The financial support of L'Association Française contre les Myopathies (AFM) and the European Commission (EU) are also acknowledged (VR). DR is partially supported by the GOSH Biomedical research Centre, which is also gratefully acknowledged. FM is supported by the Great Ormond Street Children's Charity and NIHR. The authors thank CERTUS Technology Associates Limited who host and maintain the database.
References
Footnotes
Collaborators NorthStar Clinical Network—FM, AYM, S Robb, Dr R Quinlivan, Dr A Sarkozy, VR, MM, J Butler, Dubowitz Neuromuscular Centre, Great Ormond Street Hospital for Children NHS Trust, London; Prof K Bushby, Prof V Straub, Dr M Guglieri, Dr M Eagle, AM, Institute of Human Genetics, Newcastle; Dr H Roper, H McMurchie, Birmingham Heartlands Hospital; Dr A Childs, Dr K Pysden, L Pallant, Yorkshire Regional Muscle Clinic, Leeds General Infirmary; Dr S Spinty, Dr G Peachey, A Shillington, Alder Hey Hospital, Royal Liverpool Children's NHS Trust, Liverpool; Dr E Wraige, Dr H Jungbluth, J Sheehan, R Spahr, Evelina Children's Hospital, St Thomas’ Hospital, London; Dr I Hughes, E Bateman, C Cammiss, Royal Manchester Children's Hospital, Manchester; Dr T Willis, L Groves, N Emery, The Muscle Clinic, Robert Jones & Agnes Hunt, Orthopaedic & District Hospital NHS Trust, Oswestry; Dr P Baxter, M Senior, E Scott, Sheffield Children's Hospital NHS Trust, Sheffield; Dr L Hartley, B Parsons, University Hospital of Wales, Cardiff; Dr A Majumdar, L Jenkins, B Toms, Frenchay Hospital, Bristol; Dr K Naismith, A Keddie, Armistead Child Development Centre, Kings Cross Hospital, Dundee; Dr I Horrocks, M Di Marco, Royal Hospital for Sick Children, Yorkhill, Glasgow; Dr G Chow, A Miah, Queens Medical Centre, University Hospital, Nottingham; Dr C de Goede, Preston Royal Hospital; Dr N Thomas, M Geary, J Palmer, Southampton General Hospital; Dr C White, K Greenfield, Morriston Hospital, Swansea; I Wilson, NorthStar coordinator, Dubowitz Neuromuscular Centre, Great Ormond Street Hospital for Children NHS Trust, London. Italian Neuromuscular Network—Dr S. Messina, Department of Neurosciences, University of Messina and Centro Clinico Nemo Sud, Messina; Dr A. Berardinelli, Child Neurology and Psychiatry Unit, “C. Mondino” Foundation, Pavia; Dr G. Comi, Dino Ferrari Centre, Neuroscience Section, Department of Pathophysiology and Transplantation (DEPT), University of Milan, Neurology Unit, Ca’ Granda Ospedale Maggiore Policlinico, Milan; Dr A. D'Amico, Dr E. Bertini, Department of neurosciences, unit of neuromuscular and neurodegenerative disorders, Bambino Gesù Children’s Hospital, Rome; Dr C. Bruno, Center of Myology and Neurodegenerative Disease", Istituto Giannina Gaslini, Genova; Dr L. Politano, Cardiomyology and Medical Genetics, Department of Experimental Medicine, Second University of Naples; Dr R. Battini, Department of Developmental Neuroscience, Stella Maris Institute, Pisa; E. Pegoraro, Department of Neurosciences, University of Padua, Padua; A. Pini Child, Neurology and Psychiatry Unit, IRCCS Istituto delle Scienze Neurologiche di Bologna; T. Mongini, Department of Neurosciences ‘Rita Levi Montalcini’, University of Torino; L. Morandi, Developmental Neurology Unit, Istituto Neurologico “Besta” Milan.
Contributors VR, EM and FM oversaw the design and analysis of the study. VR, MP, AM, MM, AYM, EM and FM oversaw and contributed to data collection. VR and DAR analysed the data. VR wrote the first draft of the manuscript, DAR, FM and EM contributed to the writing of the manuscript. VR, MP, AM, MM, AYM, EM and FM contributed to the revision of the manuscript.
Funding The NorthStar Clinical Network and database is supported and substantially funded by the Muscular Dystrophy Campaign, UK (Grant number: MC4/1062/4).42
Competing interests FM has served on scientific advisory boards for AcceleronPharma, Genzyme, AVI BioPharma, the Debiopharma Group, GlaxoSmithKline, Prosensa, Servier, Summit and Santhera Pharmaceutical, receives research support from Trophos and GlaxoSmithKline, and has received funding for trials from AVI and PTC. EM is site PI for the PTC extension study of Ataluren in DMD, for the TROPHOS clinical trial on SMA and for the GSK study on exon skipping. He also receives funds from the Italian Telethon and SMA Europe. He has acted as a board advisor for Shire, PTC Therapeutics and Prosensa.
Ethics approval A full application for multicentre ethical approval was made in 2006. However, it was the view of the Multicentre Research Ethics Committee that ethical approval was not required as this was a prospective audit. Approval of the study was given by the Caldicott Guardian and audit committee of each hospital Trust involved in the study. The Ethical committee from Catholic University, Rome approved this procedure for the Italian clinical network.
Provenance and peer review Not commissioned; externally peer reviewed.