Article Text

Abstract
Multiple sclerosis (MS) and neuromyelitis optica (NMO) are autoimmune demyelinating diseases of the central nervous system (CNS), having distinct immunological and pathological features. They have two pathogenic components, ‘inflammation’ and ‘neurodegeneration’, with different degrees of severity and pathogenetic mechanisms. The target antigen of autoimmunity in NMO is the water channel aquaporin-4 (AQP4), and antibodies directed against this antigen result in astrocyte damage. MS is a disease primarily affecting myelin and oligodendrocytes, but thus far, no MS-specific autoantigen has been identified. The distinct inflammatory processes in these diseases may trigger cascades of events leading to disease-specific neurodegeneration. Damage of the CNS tissue appears to be amplified by mechanisms that are in part shared by the two conditions and involve oxidative burst activation in microglia/macrophages, mitochondrial damage and axonal energy failure, Wallerian degeneration and meningeal inflammation. However, they appear to differ regarding the nature of the inflammatory response, the type and extent of cortical injury, and the type of astrocyte reaction and damage. Here, we provide a detailed comparison of the pathology between MS and NMO, which may help to define shared and disease-specific mechanisms of neurodegeneration in these diseases.
- MULTIPLE SCLEROSIS
- NEUROPATHOLOGY
- NEUROIMMUNOLOGY
Statistics from Altmetric.com
Introduction
Multiple sclerosis (MS) and neuromyelitis optica (NMO) spectrum disorders (NMOSD) are chronic inflammatory demyelinating diseases of the central nervous system (CNS) typically following a relapsing and remitting course. NMO is an autoimmune channelopathy/astrocytopathy that targets the water channel aquaporin-4 (AQP4) on astrocytes in the CNS,1 and AQP4 antibody, which has facilitated the distinction from MS (oligodendrocytopathy), is a serum diagnostic biomarker for NMO.2 However, the target antigens or specific diagnostic biomarkers have remained elusive in MS, although MS has traditionally been considered to be an autoimmune disorder mediated by aberrant T-cell and B-cell attacks against CNS, particularly myelin or oligodendrocytes.3 ,4
Both diseases occur on a chronic inflammatory reaction in the CNS, but the patterns of tissue injury and neurodegeneration are distinctively different, suggesting different mechanisms of brain injury. Here, we summarise and review the current views on the pathology and mechanisms of inflammation and neurodegeneration in MS and NMO, to provide further insights into the pathogenesis by a direct comparison between these two diseases.
Neurodegeneration in MS
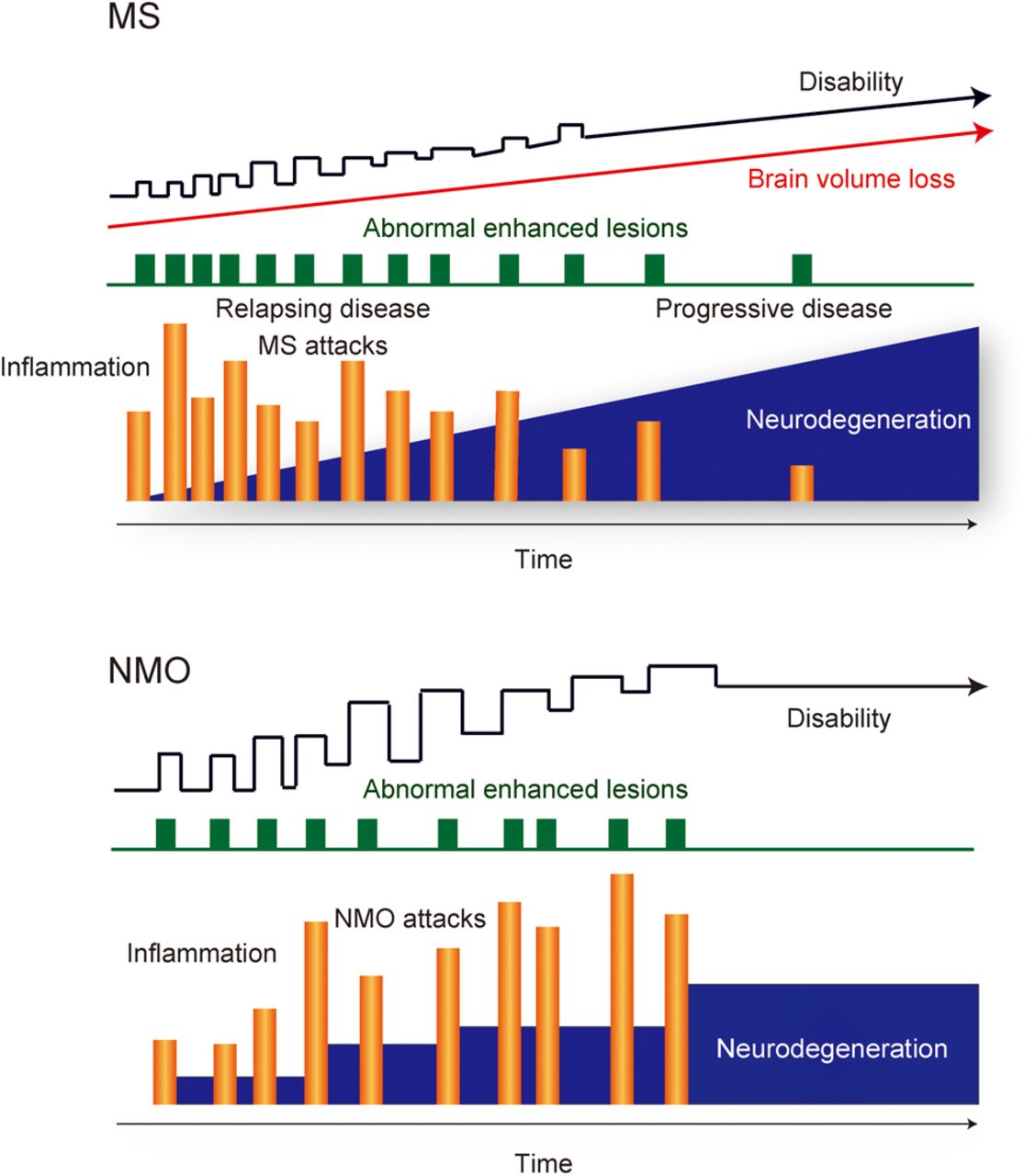
MS is one of the world's most common neurologic disorders, and the estimated number of patients with MS increased from 2.1 million in 2008 to 2.3 million in 2013.5 Recent data from natural history studies and clinical trials have shown that the interplay between ‘immunological’ and ‘neurodegenerative’ processes of MS plays an important role for entire disease courses including the relapsing–remitting course in early phases and the progressive course with physical and cognitive impairments in late phases6 ,7 (figure 1).
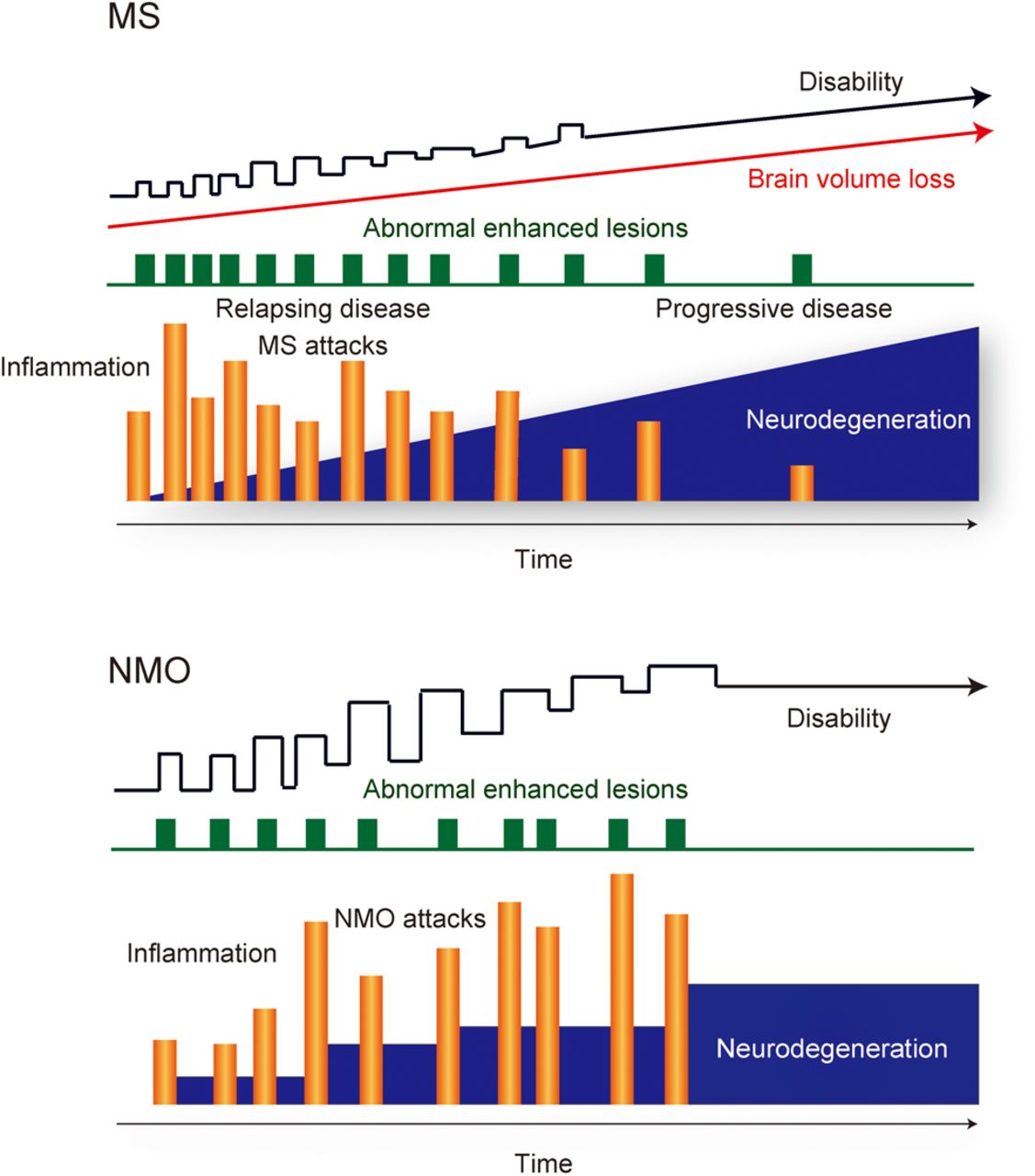
Disease courses of multiple sclerosis (MS) and neuromyelitis optica (NMO). MS and NMO are chronic inflammatory demyelinating diseases of the central nervous system typically following a relapsing and remitting course. Natural history of MS progression is largely independent of inflammatory relapse activity, and it is inconsistent with the notion that inflammation is only the sole driver of accumulating disability and neurodegeneration in progressive MS. However, progressive disease stage is only rarely seen in NMO, and neurodegeneration in NMO may be completely dependent on inflammatory relapses. They have two important pathogenic components, ‘inflammation’ and ‘neurodegeneration’, with different degrees of severity and pathogenetic mechanisms.
Clinical observations have repeatedly shown that the natural history of MS progression is usually independent of relapse activity with inflammation,8 and they are inconsistent with the idea that inflammation is the sole power of accumulating disability and degeneration in progressive MS.7 Experiences from clinical treatment trials have provided important insights into MS pathogenesis. In particular, clinical trials with disease-modifying drugs (DMDs) that target single molecules or defined pathways of the immune system (eg, natalizumab, alemtuzumab, ocrelizumab/rituximab or fingolimod) are most informative regarding molecular pathogenesis of MS in comparison to other DMDs that operate through a pleiotropic action (eg, glatiramer acetate, interferon or dimethyl fumarate).6 ‘Natalizumab’9 blocks the adhesion molecule VLA-4 and inhibits the migration of lymphocytes across the blood–brain barrier (BBB) to the CNS. ‘Alemtuzumab’10 depletes CD52+ lymphocytes, expressed not only on CD4+ T cells but also on other T-cell populations and B cells, in the peripheral immune compartment. ‘Ocrelizumab’11 or ‘rituximab’12 depletes CD20+ B cells. ‘Fingolimod’13 acts as a functional antagonist of the sphingosine-1-phosphate receptor, precludes migration of CCR7+ central memory T cells into the CNS and also exerts effects on B-lymphocyte migration and function.14 ,15 For relapsing–remitting MS (RRMS), they target defined pathways of the immune system, rapidly decrease CNS inflammation and reduce relapse rates via direct targeting of single molecules in the immune system. However, alemtuzumab16 or natalizumab17 in secondary progressive MS (SPMS) and fingolimod in primary progressive MS (PPMS)18 have little effect on disease progression, although they suppress inflammation, defined by gadolinium enhancement in MRI. Meanwhile, anti-CD20 antibodies might have some effects for progressive MS. No overall effects of rituximab, the chimeric (mouse–human) anti-CD20 antibody, was noted in patients with PPMS with confirmed progression,19 and individuals with RRMS can evolve to SPMS despite ongoing rituximab treatment.20 However, a beneficial effect of rituximab was noted in the inflammatory subgroup in patients with PPMS with gadolinium enhancing lesions at baseline.19 Ocrelizumab, a fully humanised anti-CD20 antibody, binds to epitopes on CD20, which are different compared to those targeted by rituximab, and induces stronger antibody-dependent cell-mediated cytotoxicity (ADCC) and less complement-dependent cytotoxicity (CDC). Ocrelizumab treatment met its primary end points in PPMS, showing the reduced risk of progression of clinical disability and the reduced rate of whole brain volume loss (ClinicalTrials.gov NCT01194570. National Library of Medicine. Available at: https://clinicaltrials.gov/ct2/show/NCT01194570. Results were presented at the 31st congress of the European Committee for Treatment and Research in Multiple Sclerosis in October 2015. Available at: http://www.roche.com/media/store/releases/med-cor-2015-10-08.htm).21 The different outcomes in the trials of rituximab versus ocrelizumab for PPMS might be due to differences in trial designs, characteristics of the drugs or the statistical power of the respective trials (732 patients in the ocrelizumab phase III and 439 patients in the rituximab phase II/III trials). However, despite the statistically significant effects of anti-CD20 antibody treatment (ocrelizumab) in clinical trials for PPMS, there is still substantial disease progression in the respective patients. Several potential explanations have been provided for the lack of efficacy of anti-inflammatory treatments in progressive MS:7 ,22 ,23 First, inflammation may in part be trapped behind a normal or repaired BBB in progressive MS, and systemically applied antibodies may not reach sufficient concentrations in the CNS for an effective depletion of the target cells. Second, inflammation may trigger a cascade of pathogenetic events, resulting in neurodegeneration, which no longer depends on the presence of immune cells in the CNS. Finally, neurodegeneration in MS may be a primary event, which is amplified or modified by the inflammatory response.
The inflammatory processes in MS may differ between the early (relapsing) and the progressive phases. A peripheral immune response targeting the CNS appears to drive the disease process during the early phase, whereas immune reactions within the CNS dominate during the progressive phase.6 Key immunological elements that trigger and amplify neurodegeneration in MS include the innate and adaptive immune responses, involving T cells, B cells, macrophages and dendritic cells, and microglia. However, this view has been challenged by the observation that initial stages of new plaques in MS show surprisingly little tissue infiltration by T lymphocytes and that evidence for antibody-mediated tissue injury is observed only in a subset of patients with MS.24 In this context, it has to be emphasised that inflammation (mainly consisting of perivascular inflammatory infiltrates with T and B cells and a diffuse infiltration of the CNS parenchyma by CD8+ T cells) is invariably present, even in the earliest stages of plaque formation.25
Since Charcot described that the hallmarks of MS pathology, inflammation, demyelination and relative preservation of axons are known to be key features of the disease,26 ,27 MS has for long been considered an autoimmune disease that primarily affects the myelin sheaths in white matter (WM). However, Charcot had already noticed axonal alterations including thin axons and large axonal swelling in MS plaques and the association between axonal loss and clinical disability.26 ,27 WM demyelination by itself cannot elucidate all of the cognitive impairments and physical disabilities in patients with MS. Currently, renewed interest in the pathogenetic mechanisms of neurodegeneration in WM22 ,28 and grey matter (GM)22 ,28 ,29 is emerging in MS.
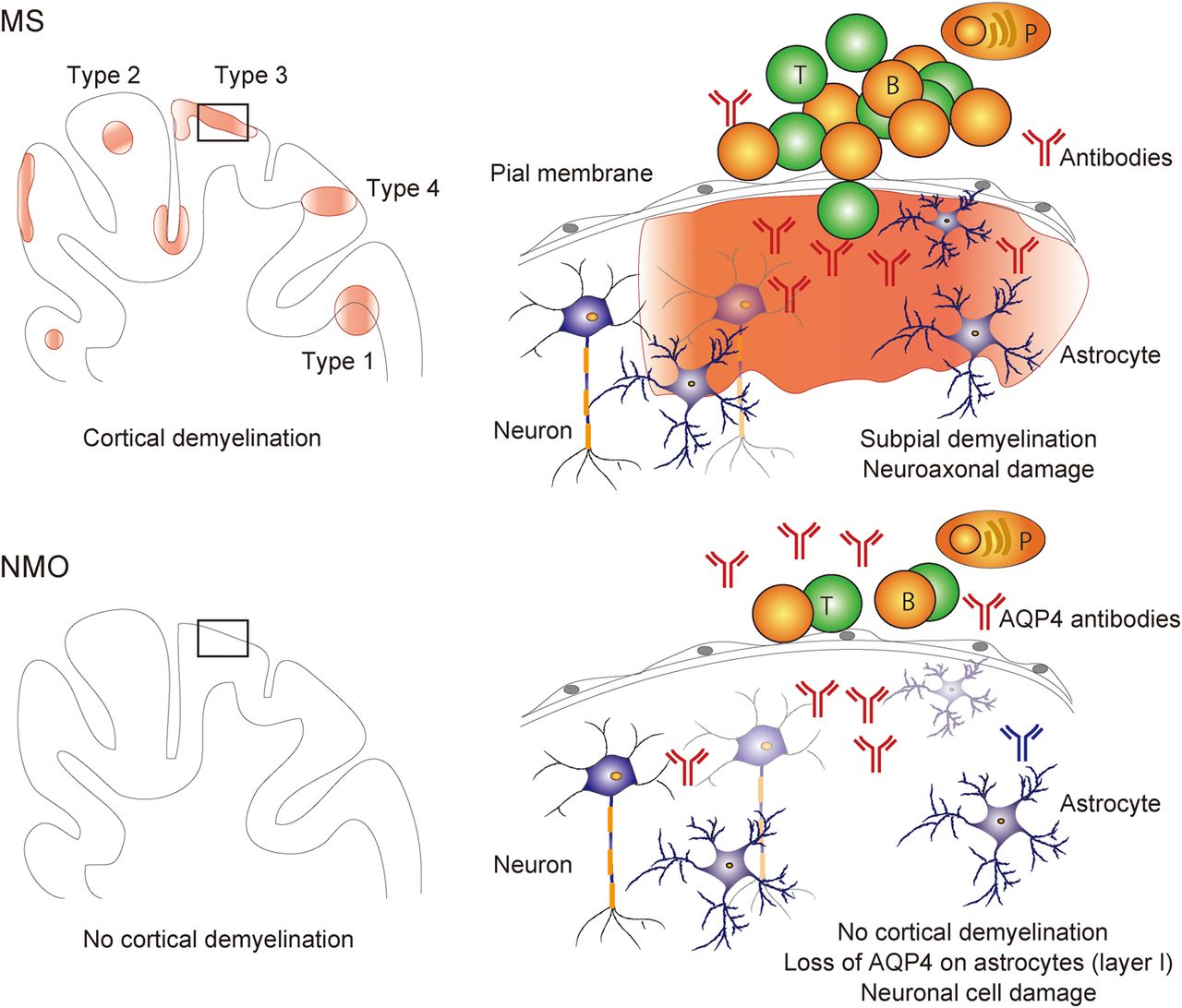
Demyelination and neurodegeneration of cortical and deep GM are present in the MS brains. Importantly, radiological assessments demonstrated that not only WM lesions but also cortical and deep GM lesions are associated with cognitive impairment30–33 and disability progression.32–35 Demyelinated cortical GM lesions are categorised into four types29 ,36–39 (figure 2): lesions that extend through WM and GM (lesion type 1, 14%):36 lesions that are located within the cortical GM and do not extend to the surface of the brain (lesion type 2, 1%);36 lesions that are located in the subpial cortex (lesion type 3, 67%);36 and lesions that extend throughout the full width of the cerebral cortex without affecting WM (lesion type 4, 17%).36 Types 2–4 are exclusively intracortical. Classically, cortical GM demyelinated lesions are pathological hallmarks of patients with PPMS and SPMS.40 It is also present in 38% of biopsy samples in a cohort of patients with early-stage MS and clinically isolated syndrome (CIS),29 although this cohort29 mainly contains patients with aggressive disease and large subcortical lesions, which may in part explain the high incidence of cortical lesions in this study in comparison to studies on autopsy tissues. Interestingly, type 3 subpial lesions, which influence the widespread cortical regions and are seen in deep invaginations of the brain surface, are related to meningeal inflammation29 and show some amounts of neuronal and axonal injuries.23 These data suggest that cytokines, specific autoantibodies or other molecular mediators produced by lymphocytes and macrophages in the meningeal aggregates may diffuse from the subarachnoid space into the underlying cortex, and then an increasingly inflammatory milieu may elicit subsequent widespread demyelination and/or neuronal loss23 ,29 ,41 (figure 2). Further radiological studies using more advanced and sensitive techniques and their postmortem verification are needed to confirm in vivo clinical evidence that cortical GM lesions are related to disease progression of physical disabilities and cognitive impairments in patients with MS. 3D double inversion recovery (DIR) sequence MR (1.5T) imaging detected only 18% of the total cortical lesions (eg, 7% of type 3 subpial cortical lesions) that were determined by pathological assessments of postmortem MS brains.42
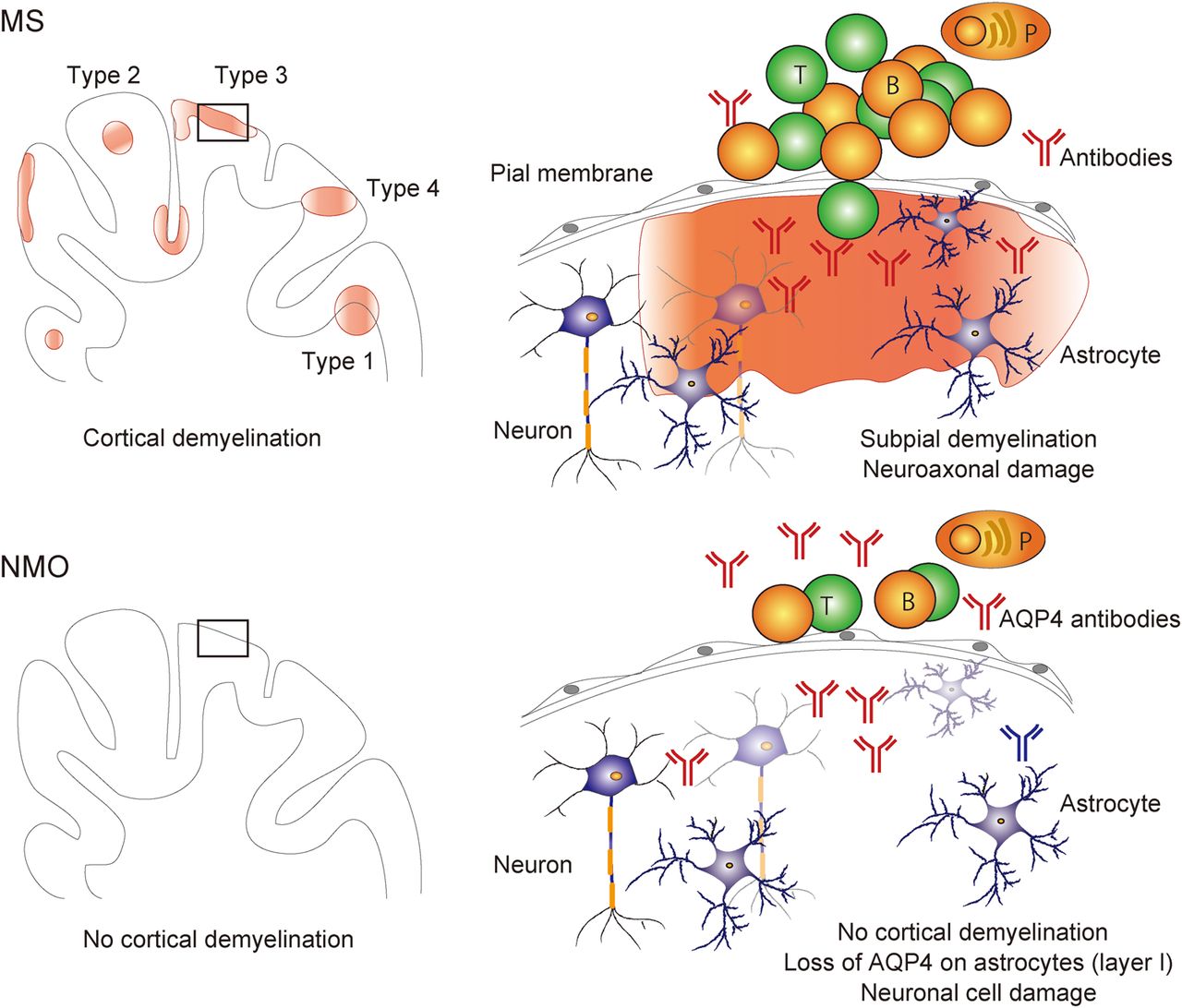
Cortical grey matter (GM) pathology of multiple sclerosis (MS) and neuromyelitis optica (NMO). Pathological processes of cortical GM in MS are distinct from those of NMO. The most important point is that demyelination in cortical GM and ectopic B-cell follicle-like structures in the meninges are evident in MS (types 1–4), but not in NMO. The cortical GM lesions in MS are characterised by demyelination, subtle neuroaxonal degeneration, transection of neurites and synaptic loss, dependent on meningeal inflammation. On the other hand, neuronal loss in cortical layers II–IV with non-lytic reaction of aquaporin-4 (AQP4)-negative astrocytes in layer I, massive activation of microglia in layer II and no deposition of complements in cortical GM of the NMO brains. T, T lymphocytes; B, B lymphocytes; P, plasma cells.
A pathological study using postmortem samples demonstrated that deep GM demyelinated lesions are also evident in patients with MS.43 They are widely seen in the MS brains but are most commonly located in the thalamus, hypothalamus and caudate nuclei. The deep GM in MS is affected by two different processes: focal demyelinated plaques and diffuse neurodegeneration.43 Both are associated with oxidative injury. These pathological data may be consistent with radiological findings indicating that thalamic atrophy is present in radiologically isolated syndrome,44 CIS45 and MS.46 Furthermore, unique GM pathology is evident in the hippocampus and the cerebellum.47 ,48 Not only minimal neuronal loss but also profound reduction in synapse density has been shown in the demyelinated lesions of the hippocampus.47 Levels of proteins that help maintain synapses and glutamate homeostasis are significantly decreased in demyelinated lesions of the hippocampus in the MS brains.47 Cerebellar cortical demyelination occurs mainly in a band-like manner, affecting multiple folia, and on average 38.7% of the cerebellar cortical area is involved.48
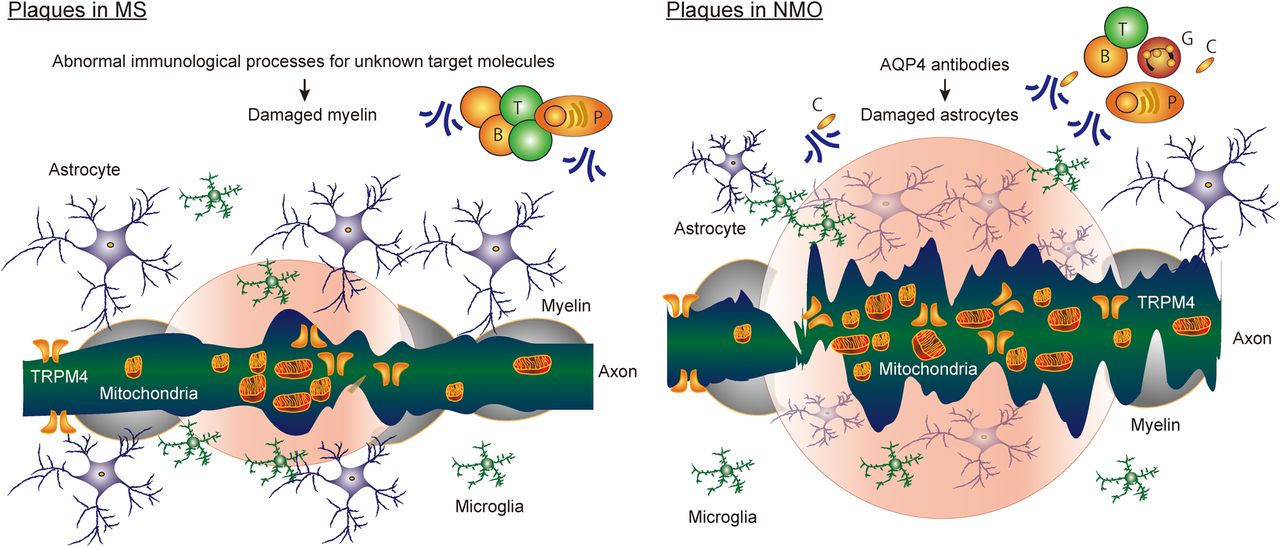
Axonal degeneration early in the disease course of MS begins in active MS lesions but does not initially cause permanent clinical disability because the brain has a remarkable ability to compensate for neuroaxonal loss.22 ,49 ,50 Gradually, accumulation of axonal loss progresses and contributes substantially to brain atrophy and neurological disability including physical and cognitive impairments during progressive MS.51 This is also reflected by changes in brain parenchymal fraction and brain volume over time in imaging studies or by the reduction of retinal nerve fibre layer (RNFL) thickness seen in optical coherence tomography (OCT).52 Furthermore, the effects of DMDs on brain atrophy correlate with treatment effect on disability in RRMS.53 The accumulation of axonal loss in all stages of MS is located not only in acute or chronic demyelinating lesions but also in normal-appearing WM and GM23 ,40 ,54 ,55 and is associated with microglia activation. Key elements driving neurodegeneration in MS are considered to be (1) oxidative burst activation of microglia/macrophages, (2) mitochondrial damage and (axonal) energy failure, (3) histotoxic (virtual) hypoxia and genuine hypoxia, (4) Wallerian degeneration (Wld), (5) age-related iron accumulation, (6) meningeal inflammation in the human brain and (7) activation of astrocytes22 ,24 ,29 ,56 ,57 (figure 3).
Oxidative activation and microglia/macrophages: Microglia and macrophages in active MS lesions and in normal-appearing WM and GM express molecules involved in the production of reactive oxygen species such as nicotinamide adenine dinucleotide phosphate oxidase and myeloperoxidase.58 More than 80% of the MS-specific genes by genome-wide microarray analysis in micro-dissected active cortical MS lesions were related to inflammation mediated by T cells, microglia activation, oxidative injury, DNA damage and repair, remyelination and regenerative processes.59 These data suggest that the inflammation-associated oxidative burst in activated microglia and macrophages plays an important role in demyelination and free radical-mediated tissue injury in the pathogenesis of MS.58
Mitochondria and axonal energy failure: Mitochondria are highly susceptible in conditions of oxidative injury.22 Mitochondrial dynamics is an important contributor to damage and loss of axons in several neurodegenerative disorders.60 Axons are extremely susceptible to mitochondrial dysfunction because the geometry of the neuron–axon unit presents a substantial challenge for efficient distribution of mitochondria and ATP production within axons, which can reach 1 m or more in length in humans.22 ‘Fusion and fission’ and an ‘increased number’ of mitochondria play critical roles in maintaining functional mitochondria in the presence of environmental stress.60 These dynamics of mitochondria fine-tune fundamental cellular processes such as calcium homeostasis and the generation of ATP and reactive oxygen species and consequently play important roles in apoptosis, mitophagy and oxygen sensing.60 Intriguingly, in the mouse experimental autoimmune encephalomyelitis (EAE) model, focal axonal degeneration/swelling with focal intra-axonal accumulation of dysmorphic and swollen mitochondria, but with intact myelin sheaths, is the earliest ultrastructural sign of damage, which progresses to axonal fragmentation and demyelination.61 These axonal structural changes that are consistent with focal axonal degeneration/swelling are present in acute MS lesions,61 in demyelinated axons of chronic inactive MS lesions62 or in remyelinated axons in the MS brain.62 Lack of mitochondrial respiratory chain complex IV activity in a proportion of Na+/K+ ATPase α-1+-demyelinated axons supports axonal dysfunction as a contributor to neurological impairment in MS.63 Furthermore, in neurons of the cortical GM, multiple deletions of mitochondrial DNA (mtDNA) were apparent in MS. The respiratory-deficient neurons harboured high levels of clonally expanded mtDNA deletions at a single-cell level, and there were neurons lacking mtDNA-encoded catalytic subunits of complex IV.64 These data suggest that neurons in MS are in part respiratory-deficient due to mtDNA deletions, which are extensive in GM and are most likely induced by inflammation. Multiple deletions of mtDNA contribute to neurodegeneration in MS.64 Moreover, a significant reduction of peroxisome proliferator-activated receptor gamma coactivator-1α, a transcriptional coactivator, leads to reduced transcription of mitochondrial respiratory chain complex subunits and mitochondrial antioxidants in normal-appearing GM samples of progressive MS.65 Overall, mitochondrial injury in MS results in energy deficiency, respiratory chain dysfunction, oxidative injury and altered calcium homeostasis, causing a vicious circle in the process of tissue destruction22 (figure 3).
Virtual hypoxia and genuine hypoxia: Hypoxia plays an important role in processes of neurodegeneration in MS, based on the following evidences. First, distal oligodendrogliopathy, characterised by degeneration of the most distal oligodendrocyte processes, the selective loss of myelin-associated glycoprotein from affected myelin sheaths and apoptotic destruction of oligodendrocytes,66 is seen not only in all patients within the first days after initiation of a WM stroke lesion but also in patients with MS with pattern III lesions in early and active disease stages. Second, MS lesions with activated microglia and macrophages in chronic demyelinated axons are associated with mitochondrial damage, oxidative injury, electron leakage and iron liberation from oligodendrocytes during demyelination, resulting in a stage of energy deficiency or ‘virtual hypoxia’.67 Key alterations of the ‘virtual hypoxia’ might include mitochondrial dysfunction, Na+ influx through voltage-gated Na+ channels and axonal AMPA receptors, release of toxic Ca2+ from the axoplasmic reticulum, over-activation of ionotropic and metabotropic axonal glutamate receptors, and activation of voltage-gated Ca2+ channels, ultimately leading to excessive stimulation of Ca2+-dependent degradation pathways.67 Third, ‘genuine hypoxia’ also plays an important role in neurodegeneration in MS. Focal demyelinated lesions in the WM occurred not only at sites with high venous density but also in watershed areas of low arterial blood supply.56 Moreover, the experimental model using microinjection of lipopolysaccharide into the rat spinal dorsal column indicated that demyelination characteristic of at least some early MS lesions can arise at a vascular watershed following activation of innate immune mechanisms that provoke hypoxia, and superoxide and nitric oxide formation. Demyelination in this model can be reduced or prevented by increasing inspired oxygen to alleviate the transient hypoxia.68 It is possible to hypothesise that the ‘neurovascular unit’, which includes neurons, non-neuronal cells such as vascular cells (endothelia, pericytes and vascular smooth muscle cells) and glia, plays a major role in the disease pathogenesis of various neurological disorders, including MS, Alzheimer's disease or amyotrophic lateral sclerosis (ALS). It maintains the chemical and cellular composition of the neuronal milieu, which is required for proper functioning of neuronal synapses and circuits as well as for remodelling, angiogenesis and neurogenesis.69
Wallerian degeneration (Wld): Wld, which was traditionally thought to result from passive degeneration of axons due to a lack of cell body-derived nutrients, is a prominent early feature of most neurodegenerative disorders, including ALS. However, analysis of the slow Wallerian degeneration (WldS) mutant mouse provided evidence that it is mediated by an active auto-destruction programme involving axon death signalling pathways, including the Toll-like receptor adaptor SARM1.70 In the early stages of Wld, mitochondria swell, fragments of mitochondria accumulate at paranodes and mitochondria lose their membrane potential.70 In the EAE model, WldS-mutant mice show a modest attenuation of axonal loss, suggesting that axonal damage may occur by a mechanism that is similar to Wld.71 In the early disease course of MS, Wld is a major element of neuroaxonal pathology in plaques and periplaque WM.72
Age-related iron accumulation: Oxidative injury is amplified in the presence of divalent cations such as iron (Fe2+) or copper (Cu2+), and iron accumulates within the normal human brain in an age-dependent manner.22 Iron decreases in the normal-appearing WM of patients with MS with increasing disease duration, and cellular degeneration in MS lesions leads to waves of iron liberation, which may propagate neurodegeneration together with inflammatory oxidative burst.22
Meningeal inflammation: Meningeal inflammation is prominent in all MS courses including even early MS and is topographically associated with cortical demyelination.29 ,56 Incidence and size of demyelinated lesions are significantly larger in cortical sulci, and in deep invaginations of the brain surface, such as the cingulate gyrus and in the insular cortex.56 The topographical distribution of meningeal inflammatory infiltrates, which are most prominent in cerebral sulci and in deep invaginations of the brain surface, is significantly associated with subpial cortical demyelination.56 Moreover, ectopic lymphoid neogenesis can be present in leptomeninges of the MS brain. It has been reported that ectopic lymphoid neogenesis is evident in the target organ of several autoimmune disorders:73 joints of rheumatoid arthritis, the thyroid gland of Hashimoto's thyroiditis, the salivary glands of Sjögren's syndrome, the pachymeninges of myeloperoxidase antineutrophil cytoplasmic antibody-positive hypertrophic pachymeningitis74 and the nasal passages and lungs of patients with granulomatosis with polyangiitis. The presence of organised lymphoid structures that in part resemble lymphoid follicles in chronically inflamed tissues indicates that lymphoid neogenesis might have a role in maintaining immune responses against persistent antigens.73 Recently, a clear gradient of neuronal loss became evident not only in GM demyelination but also in normal-appearing GM when accompanied by meningeal B-cell follicle-like structures.23 These data indicate that some neurodegeneration in the cortex may be independent of demyelination and may occur more in a diffuse manner.23
Activation of astrocytes: Astrocytes are the most abundant cells in the CNS, and they may regulate the activity of microglia, oligodendrocytes and cells of the adaptive immune system under neuroinflammation.57 Lactosylceramide (LacCer) is synthesised by β-1,4-galactosyltransferase 6 (B4GALT6) and acts in an autocrine manner to control astrocyte transcriptional programmes. It is upregulated in the CNS in the rodent model of chronic progressive EAE and in lesions of human MS. LacCer in astrocytes controls the recruitment and activation of microglia and CNS-infiltrating monocytes in a non-cell autonomous manner by regulating production of the chemokine CCL2 and granulocyte-macrophage colony-stimulating factor, respectively. Inhibition of LacCer synthesis in mice suppressed local CNS innate immunity and neurodegeneration in EAE. These data suggest that B4GALT6 regulates astrocyte activation and is a potential therapeutic target for progressive MS.57 Moreover, although astrocyte glycogenesis and lactate metabolism function to maintain brain activity in normal conditions, astrocytes are deficient in β2 adrenergic receptors in the MS brain, resulting in decreased formation of cAMP, subsequently a decrease in glycogenolysis, and a decreased production of lactate, leading to impaired axonal mitochondrial metabolism and axonal degeneration.75 ,76
{kind=link}
{kind=link}
{kind=link}
Models of axonal degenerative pathology of multiple sclerosis (MS) and neuromyelitis optica (NMO). Key elements driving neurodegeneration in MS are considered to be oxidative burst activation of microglia/macrophages, mitochondrial damage and axonal energy failure, histotoxic (virtual) hypoxia and genuine hypoxia, Wallerian degeneration, age-related iron accumulation, accumulation of transient receptor potential melastatin 4 channel (TRPM4) and meningeal inflammation in the human brain. Meanwhile, a severe phenotype of NMO can be explained by profound tissue damage including neuroaxonal injury, occurring as a secondary consequence of astrocyte damage by aquaporin-4 (AQP4) antibodies and more severe neurodegeneration, including axonal accumulation of damaged mitochondria and TRPM4 in NMO acute lesions. However, it is not yet finally proved that chronic and diffuse neurodegeneration is involved in the NMO brain. T, T lymphocytes; B, B lymphocytes; P, plasma cells; C, activated complements.
Neurodegeneration in NMO
NMO is recognised as an autoimmune and inflammatory CNS disorder with seropositivity of AQP4 antibodies.1 It is a distinct disease from MS with respect to the immunological processes, clinical disease courses and treatment responses.2 ,77 ,78 Lesions of the spinal cord in NMO are characterised by the presence of longitudinally extensive damage with central GM involvement.2 ,77 ,78 Lesions of the visual pathway in NMO often show severe and bilateral involvement, and are accompanied by thinning of an RNFL by OCT.2 ,77–80 Area postrema lesions of NMO often give rise to intractable vomiting, nausea or hiccups.2 ,77 ,78 ,81 Furthermore, brainstem lesions, cerebellar lesions, extensive WM lesions82 ,83 or cortical GM lesions84 are seen in some patients with NMO. Brain abnormalities on MRI are present in 60% of patients with NMO,82 and 5–42% of the patients fulfil the Barkhof criteria for MS.83 ,85
AQP4 water channels as an autoantigen in patients with NMO1 are abundant in foot processes of astrocytes at the BBB. The key characteristics of NMO pathology are loss of AQP4 immunoreactivity on astrocytes with vasculocentric deposition of immunoglobulins and activated complement86–88 and infiltration of inflammatory cells, including lymphocytes, plasma cells89 and granulocytes.88 These findings are similar in the definite and limited form of NMO (NMOSD).90 Previous studies including rodent models have demonstrated that CDC,91 ADCC92 and internalisation of AQP4 channels93 might fulfil prominent tasks in pathogenesis of NMO, although internalisation of AQP4 channels is a controversial issue.94 Reflecting these mechanisms, NMO lesions are pathologically characterised by six types.95 Type 1: active lesions associated with complement activation and granulocyte infiltration. Type 2: necrotic and cystic lesions with extensive tissue destruction. Type 3: secondary Wld in lesion-associated tracts. Type 4: lesions with selective loss of AQP4 in the absence of other structural damage. Type 5: active lesions with clasmatodendrosis of astrocytes, defined by cytoplasmic swelling and vacuolation, beading and dissolution of their processes and nuclear alterations resembling apoptosis. This may be associated with internalisation of AQP4 and astrocyte apoptosis in the absence of complement activation. Type 6: lesions with astrocyte dystrophy and primary demyelination.95
Involvement of the optic nerves and the spinal cord in NMO is associated with severe visual impairment and poor prognosis of optic neuritis79 or more severe motor/sensory disabilities for myelitis, in comparison to the respective changes seen in MS. A severe phenotype of NMO is associated with profound tissue damage including neuroaxonal injury, occurring as a secondary consequence of astrocyte damage by AQP4 antibodies. Related to optic neuritis as a representative NMO lesion, the pathological correlates of the severity in NMO have been provided as follows:79 (1) Longitudinally extensive optic neuritis. (2) Unique dynamics of AQP4, including (i) loss of AQP4 immunoreactivity on astrocytes and complement activation in optic neuritis lesions, (ii) loss of AQP4 immunoreactivity on Müller cells and no complement activation and (iii) densely packed AQP4 immunoreactivity on astrocytes in gliosis at sites of secondary anterograde/retrograde degeneration in the optic nerves and RNFL. (3) More severe neurodegenerative processes with accumulation of damaged mitochondria and transient receptor potential melastatin 4 channel (TRPM4) in axons with astrocyte pathology via CDC in optic neuritis lesions.79 RNFL thinning and loss of ganglion cells in the presence of AQP4+ astrocytes indicate secondary retrograde degeneration after optic neuritis.79 Furthermore, in vivo imaging of the spinal cord to study the formation of experimental NMO-related lesions caused by human AQP4 antibody in mice demonstrated that human AQP4 antibody results in acute depletion of astrocytes with initial oligodendrocyte survival.96 This was followed after 2 hours of antibody application by secondary axonal injury.96 Damage of astrocytes with axonal degeneration was dependent on titre of AQP4 antibody and complement. Data from this experimental rodent model using in vivo imaging96 are consistent with the patterns of neurodegeneration described in human NMO pathology79 (figure 3).
Moreover, unique lesions in cortical GM are present in the NMO brains but are different from those in the MS brains.84 ,97 Cortical GM lesions of NMO are characterised by scattered loss of cortical neurons in layers II–IV, loss of AQP4 immunoreactivity on astrocytes in layer I in the absence of complement deposition, microglia activation in layer II and meningeal infiltrations of lymphocytes.84 Importantly, no evidences of cortical demyelination or of meningeal lymphoid follicle-like infiltrates are seen in the NMO brains.84 ,97 This suggests that cortical neurodegeneration is present in the NMO brains, which is different from that seen in the MS brains, and occurs independently of CDC2 ,39 ,84 (figure 2). Neuronal loss in the GM in NMO is due in part to anterograde (Wld) or retrograde axonal degeneration following acute NMO lesions, or may be a direct consequence of the loss of trophic support following the destruction of astrocytes/Müller cells. The absence of cortical demyelination in the NMO brains in pathological studies84 ,97 is in agreement with radiological findings using ultrahigh-field MR (7T) imaging98 ,99 and DIR sequence MR (1.5T) imaging.100 Further radiological studies with more advanced and sensitive techniques are needed to investigate other pathological changes including neuronal loss in cortical layers II–IV and AQP4 loss from astrocytes in layer I84 in the NMO brains. So far, radiological studies demonstrated normal volumes and microstructural integrity of deep GM structures in the NMO brains,101 but pathological confirmation would be needed.
Neurodegeneration in AQP4-deficient regions, such as myelin-preserved periplaque WM areas, may indicate that demyelination is not essential for the neurodegenerative processes in NMO. Abnormalities of the astrocytic Na+-dependent excitatory amino acid transporter102 ,103 or downregulations of NR1 subunit of the NMDA receptor103 are present in regions with loss of AQP4 immunoreactivity in patients with NMO and in rats after passive transfer of AQP4 antibodies, and these occur even when myelin sheaths are preserved.103 They indicate that imbalances of glutamate homeostasis102 ,103 may play an important role in driving neurodegeneration via interactions between glia and neurons in NMO. Other possible elements in NMO are expected to be caused by microglia/macrophage activation,79 ,88 mitochondrial damage and axonal energy failure,79 Wld79 ,95 and meningeal inflammation79 ,84 because they are present in NMO lesions, although a direct comparison between MS and NMO, analysing the severity of these disease mechanisms, is still missing. Importantly, brain pathology in NMO, in contrast to that in MS, is characterised not just by the absence of cortical GM demyelination or of ectopic meningeal lymphoid follicle-like structures but also by the loss of astrocyte function. These differences in key pathways of neurodegeneration between NMO and MS may reflect the disease courses; a secondary progressive clinical course is extremely rare in NMO, but more than two-thirds of people with relapsing disease of MS subsequently experience secondary progression104 (figure 1). However, detailed neuropathological studies, which have addressed these potential mechanisms, have not yet been performed. Additional studies are also needed to investigate details of NMO pathogenesis, such as alteration of synaptic function, network activity or mechanisms of myelin pathology following astrocytopathy in NMO.
Conclusions and future directions
There is a major clinical difference between MS and NMO, which may shed light on mechanisms of neurodegeneration in these diseases. In MS, relapsing–remitting disease in most patients evolves into a secondary progressive course or the disease may present with a primary progressive course from the onset. This progressive disease stage is only rarely seen in patients with NMO.104 This is surprising since both diseases share the chronic inflammatory nature, they both may affect patients in the same critical age, which is associated with progressive disease conversion in MS, and they both have large focal lesions with axonal loss. Furthermore, it is likely, but not yet finally proved, that common neurodegenerative pathways, involving oxidative injury, mitochondrial damage and virtual hypoxia, are involved in tissue injury in both conditions. However, there are a number of differences in pathology and pathogenetic mechanisms, which may be relevant for the presence or absence of chronic progressive neurodegeneration.
Inflammation: Although both diseases are associated with a chronic inflammatory reaction, the nature of the inflammatory response seems to be different.89 This is further supported by the observation that some of the anti-inflammatory treatments that are highly effective in patients with MS fail in NMO or may even amplify the disease. These are type 1 interferons,105 natalizumab106 or fingolimod.107 Information on the immunological mechanisms responsible for driving the inflammatory response in MS and NMO is currently limited, and little is known about how this inflammatory response is linked to neurodegeneration.
Meningeal lymph follicle-like inflammatory aggregates: As described above, such aggregates are abundant in the MS brain, but they are largely absent in the brain of patients with NMO.84 In MS, this meningeal inflammatory process is associated with progressive cortical demyelination and neurodegeneration.56 Cortical demyelination or atrophy is associated with irreversible clinical deficit in patients with MS, including cognitive deficiencies, and meningeal inflammation correlates with the extent of active neurodegeneration in the WM as well as GM.56 ,108 Furthermore, the extent of cortical demyelination is a much better correlate for diffuse degeneration in the normal-appearing WM compared to the number, size or destructiveness of WM lesions.40 Thus, compartmentalised inflammation in the meninges may be a major driving force for disease progression in MS, and this appears to be absent in NMO.
Target cells of tissue damage: Finally, the target cells for the primary damage in the CNS are different in MS in comparison to NMO. In MS, oligodendrocytes and myelin are damaged first, whereas in NMO, the prime target is the astrocyte. Astrocyte loss may augment excitotoxic neuronal or axonal damage, but activated astrocytes can also augment brain inflammation, tissue injury and neurodegeneration.109 ,110 Thus, astrocyte loss in acute NMO lesions may play a major role in focal neurodegeneration in active NMO lesions,79 whereas chronic and diffuse astrocyte activation in the MS brain may be involved in chronic progressive neurodegeneration. On the other hand, iron in the normal ageing brain is mainly stored in myelin and oligodendrocytes. Oligodendrocyte destruction may augment oxidative injury due to the liberation of iron from the intracellular pool,111 and this may be more pronounced in a disease that primarily affects oligodendrocytes in comparison to an astrocytopathic condition. Addressing these topics in future research may provide major contributions for the understanding of the pathogenesis of neurodegeneration in these diseases.
The different neurodegenerative processes in MS and NMO are thus far only poorly represented by experimental models, and new experimental models have to be developed to understand their pathogenesis and test the effect of anti-inflammatory, neuroprotective and neuroregenerative therapies. Future therapeutic strategies for MS and NMO may not only target inflammation, but they may also include protection of axonal mitochondria,22 inhibition of TRPM4 accumulation in injured axons112 and strategies to reduce Wld-induced neurodegeneration.70
References
Footnotes
Contributors IK and HL wrote and edited the manuscript.
Funding This work was supported in part by JSPS KAKENHI grant number 26461289 (IK).
Competing interests IK has received funding for travel/speaker honoraria or for serving on scientific advisory boards from Novartis, Biogen, Bayer, Mitsubishi Tanabe, Takeda and Astellas. HL received honoraria for lectures from Novartis, Biogen, TEVA and Sanofi-Aventis.
Provenance and peer review Commissioned; externally peer reviewed.