Article Text

Abstract
Objective To investigate whether oral administration of a standardised frankincense extract (SFE) is safe and reduces disease activity in patients with relapsing-remitting multiple sclerosis (RRMS).
Methods We performed an investigator-initiated, bicentric phase IIa, open-label, baseline-to-treatment pilot study with an oral SFE in patients with RRMS (NCT01450124). After a 4-month baseline observation phase, patients were treated for 8 months with an option to extend treatment for up to 36 months. The primary outcome measures were the number and volume of contrast-enhancing lesions (CEL) measured in MRI during the 4-month treatment period compared with the 4-month baseline period. Eighty patients were screened at two centres, 38 patients were included in the trial, 28 completed the 8-month treatment period and 18 of these participated in the extension period.
Results The SFE significantly reduced the median number of monthly CELs from 1.00 (IQR 0.75–3.38) to 0.50 (IQR 0.00–1.13; difference −0.625, 95% CI −1.25 to −0.50; P<0.0001) at months 5–8. We observed significantly less brain atrophy as assessed by parenchymal brain volume change (P=0.0081). Adverse events were generally mild (57.7%) or moderate (38.6%) and comprised mainly gastrointestinal symptoms and minor infections. Mechanistic studies showed a significant increase in regulatory CD4+ T cell markers and a significant decrease in interleukin-17A-producing CD8+ T cells indicating a distinct mechanism of action of the study drug.
Interpretation The oral SFE was safe, tolerated well and exhibited beneficial effects on RRMS disease activity warranting further investigation in a controlled phase IIb or III trial.
Clinical trial registration NCT01450124; Results.
Statistics from Altmetric.com
Introduction
Multiple sclerosis (MS) is a debilitating inflammatory and neurodegenerative autoimmune disease of the central nervous system that predominantly affects young women.1 It is now widely recommended that anti-inflammatory treatment be started early in the relapsing phase of the disease to prevent the development of chronic neuroinflammation and neurodegeneration.2 Several anti-inflammatory drugs have been licensed, but most of these act broadly on the immune system, and require close monitoring for risks and side effects.3 The long-term treatment of MS that is initiated early in the course of the disease therefore requires a—as yet unavailable—very well-tolerated, safe, oral immunomodulatory treatment, particularly for those patients with only mild clinical involvement and indications of a benign course.
Many patients with MS are very interested in complementary medicine (CM),4 and seek advice regarding phytotherapeutics in addition to, or even instead of, standard treatment options. Several surveys report that 70%–80% of patients with MS have used CM drugs or interventions,4 underlining the importance of CM from the patients’ perspective.
Boswellic acids (BA) are thought to be the bioactive principles of frankincense, which has been used for literally thousands of years as an anti-inflammatory substance in traditional Eastern and Oriental medicine. Several small, randomised clinical trials have shown a favourable safety and tolerability profile of frankincense extracts in a number of inflammatory and autoimmune diseases.5 The most convincing data are presented in a Cochrane review of oral herbal therapies in osteoarthritis.6 Diverse immunomodulatory mechanisms have been attributed to BAs, most prominent of which are the inhibition of the enzymes 5-lipoxygenase, microsomal prostaglandin E2 synthase-1, LL-37 and the inhibition of nuclear factor-κB activities.7–9 Previous data from our group showed that BAs interfere with CD4+ T helper cell 17 polarisation by blocking interleukin (IL)-1β signalling in vitro.10 Since involvement of several of the molecules and pathways mentioned above has been reported in the context of MS pathology,11–13 we reasoned that it would be worthwhile to assess the use of a standardised frankincense extract (SFE) as an anti-inflammatory and immunomodulatory therapeutic approach in patients with MS.
Methods
Study design and ethics statement
We performed a bicentric, phase IIa, open-label, baseline-to-treatment trial between 21st September 2011 and 7th March 2017 with an orally available SFE produced by Alpinia Laudanum Institute of Phytopharmaceutical Sciences (Walenstadt, Switzerland) at two German tertiary academic MS centres. Based on safety data and patients’ requests, an extension of the study with the option to receive the study drug for up to 36 months was applied for and approved. The trial is registered with ClinicalTrials.gov, No. NCT01450124. Participants gave their written informed consent at screening and were informed of alternative approved treatments by an independent neurologist before entering the trial. An independent data safety monitoring board consisting of three international MS experts was appointed to monitor safety and efficacy.
The trial design is presented in figure 1A. The trial consisted of four phases: baseline observation (stage 1), individual dose-finding phase (stage 2), treatment phase (stage 3) and extension (stage 4). After consenting, the patients entered a 3-month baseline observation phase (stage 1) with monthly visits at the respective study centre for contrast-enhanced brain MRI scans and clinical scoring. The SFE was provided as capsules containing 400 mg. During the first 8 weeks the patients participated in an individualised dose-finding phase (stage 2) with two parts. In part 1, up to 400 mg capsules of an SFE were used to titrate up to a maximum well-tolerated dose or to a maximum of 4800 mg/day (whichever occurred first), that is, 1600 mg three times a day in the first 28 days by adding one capsule every second or third day. After the individual maximum well-tolerated dose had been determined, the patients continued with that dose for another 28 days (part 2) for stabilisation and to assess tolerability. This was followed by 6 months of continuous treatment at this dose (stage 3). A minimum tolerated dose of 2400 mg/day was mandatory to continue with the trial. If a relapse occurred during the study, the patients were offered the option to discontinue the trial and revert to standard treatment, and an informed reconsent was necessary if they chose to continue.
- Download figure
- Open in new tab
- Download powerpoint
Trial design (A) and trial profile (B). CEL, contrast-enhancing lesion; MS, multiple sclerosis; SFE, standardised frankincense extract.
Patients were seen monthly for assessment and MRI at the study centre until month 8, then again at month 12, and afterwards at quarter-yearly intervals. Details on the inclusion and exclusion criteria and dosing regimen are provided in online supplementary tables 1 and 2 and in the study protocol.
Supplementary file 2
Study population
Eligible patients were male or female between the ages of 18 and 55 with the diagnosis of relapsing-remitting multiple sclerosis (RRMS) or clinically isolated syndrome according to McDonald criteria,14 a baseline disability score of 0–5.5 on the Expanded Disability Status Scale (EDSS),15 and an average of at least 0.5 gadolinium-enhancing MRI lesions (contrast-enhancing lesions, CEL) per month, that is, at least two CEL lesions in the four MRI scans during the pretreatment baseline period. Key exclusion criteria were liver enzyme levels more than three times the upper normal limit, serological evidence of active hepatitis B or C infection, or other chronic liver disease, a positive pregnancy test, nausea or vomiting as a frequent complaint, or known hypersensitivity to BAs or frankincense. If patients had previously been taking MS disease-modifying medication, the treatment must have been suspended for at least 12 weeks with glatiramer acetate and interferon (IFN) beta, and for 24 weeks for all other medications prior to the first investigational drug dose.
Outcome measures
The primary outcome measures were the changes in the median number of CELs and the lesion volume between baseline and treatment phases. Secondary outcome measures were the number of new active lesions (CELs, T2 lesions), amount of brain atrophy (parenchymal brain volume change, PBVC) and the annualised relapse rate (ARR), which describes the number of confirmed relapses per year. All MRI data were acquired on a 3T MRI scanner (Skyra or Trio syngo MR, Siemens Medical Systems, Germany) using a standardised protocol implemented at each site (further details in online supplementary material). Lesion masking and analysis were performed using the software Analyze V.11.0 (Analyze Direct, Biomedical Imaging Resource, Mayo Clinic, USA). MS lesions were marked as region of interest (ROI) using a semiautomated threshold-based algorithm. Lesions were outlined on each slice on which they appeared. Confluent lesions were counted as one lesion. T2-hyperintense lesions were marked on T2w images only when they exhibited a corresponding hyperintensity in the corresponding fluid attenuation inversion recovery image. On T1w images, hypointense (darker than cortical grey matter) areas were marked as black holes, only if presenting with a corresponding T2-hyperintense lesion. Finally, CELs were marked in the T1Gd images. Lesion volumes for all three lesion classes were calculated from the defined ROIs. All images were processed with the FMRIB Software Library (FSL) toolbox (FMRIB Analysis Group, University of Oxford, Oxford, UK) as described previously.16 Brain tissue volume, normalised for head size (normalised brain volume, normalised grey matter, normalised white matter), was estimated with SIENAX,17 and lesion volume was normalised based on the SIENAX results. In order to reduce the risk of false tissue assignment in lesions, the lesion masks were dilated and filled with normal appearing white matter contrast before the images were processed with SIENAX. Brain masks were manually corrected to minimise false tissue assignment by the FSL segmentation. Longitudinal atrophy was assessed with SIENAX and results were corrected for the individual duration between the four baseline scans to calculate an annualised PBVC. Tertiary outcomes comprised the EDSS,15 the Scripps Neurological Rating Scale (SNRS),18 the multiple sclerosis functional composite (MSFC)19 consisting of the Nine-Hole Peg Test, timed 25-foot walk, Paced Auditory Serial Addition Test (PASAT), the Symbol Digit Modalities Test (SDMT),20 a depression scale (Hospital Anxiety and Depression Scale21), the Fatigue Severity Scale according to22 the Hamburg Quality of Life Questionnaire23 and immunological biomarkers (lymphocyte subpopulations, cytokine levels in serum) until months 8 and 12. The trial was designed to provide class III evidence for all listed research questions. Further details for methods for immunological outcome parameters are presented in online supplementary material.
Supplementary file 1
Statistical analysis
Based on the analyses of a natural history cohort studied with monthly MRIs for a minimum of 1 year,24 we calculated that a sample size of 30 patients completing the treatment period would be necessary to detect a 40% reduction in CELs with a power of 80% and an alpha error of 0.05 (one sided). Assuming a 20% dropout rate, at least 36 patients would have to be included. We estimated that it would be necessary to screen 75 patients in order to identify 36 patients who met our inclusion criteria.
The primary and secondary endpoints are summarised as medians with IQRs. Changes from baseline to follow-up are described by their medians with 95% CIs and tested for deviation from 0 using the Wilcoxon signed-rank test as specified in the study protocol. To determine the sensitivity of the results to missing data, changes in outcome from baseline to follow-up were imputed as 0 (‘intention-to-treat analysis’). This corresponds to a baseline-carried-forward analysis, which was considered a conservative assessment. Secondary and tertiary endpoints were analysed in a similar manner. Relapses were analysed using a Poisson regression model with adjustment for overdispersion.
Results
Recruitment
We screened 80 patients and enrolled 38 of them (figure 1B). The baseline characteristics of the study population are presented in table 1. There was no significant difference in the MS demographic and disease activity markers between the patient groups considered for intention-to-treat (ITT) analysis (n=38, all patients exposed to SFE) and for primary endpoint analysis (n=28).
Baseline characteristics of the IIT and the PP patient cohort
Ten patients (26%) did not reach the primary endpoint: six patients (16%) dropped out already during the dosing phase, four patients (10%) during months 3–6. When asked for their reasons to withdraw, four patients were not able to comply with the time-consuming study protocol, four patients withdrew because of a relapse and decision to start with approved medication (two during the dosing phase, two at month 4 and month 5, respectively). One patient was not able to swallow or drink the capsules regularly (gustatory disgust) and one patient withdrew early because of a persisting urticaria.
Dosing
Thirty-four patients tolerated the maximum protocol dose of the SFE of 4800 mg/day (n=34/38; 89.5%) throughout the entire trial. In three patients, the dose had to be kept at 3600 mg SFE per day, and one patient only tolerated the minimum dose of 2400 mg/day. At month 8, the majority of the patients (26/28; 92.8%) still took the maximum dose of 4800 mg SFE daily.
Magnetic resonance imaging
The primary outcome was met for the total number of CELs as well as the volume of CELs (table 2 and figure 2A). The total number of CELs decreased significantly from a baseline median of 1.0 (IQR 0.75–3.38) to 0.5 (IQR 0.00–1.13) during the treatment period (months 5–8; P<0.0001). The volume of the CELs decreased significantly from a median of 1753.5 mm3 (IQR 553–4974.5 mm3) during the baseline phase (months −3 to 0) to 185 mm3 (IQR 0.00–1450.00 mm3; n=28; P=0.0481). The ITT analysis also gave significant results (see table 2). Both analyses showed more than 60% reduction of the total number of CELs between the baseline and treatment phase.

MRI outcomes of the SABA trial. (A) Median number of contrast-enhancing lesions (CEL) per patient detected in the monthly MRI visits. White bars indicate the baseline phase before start of treatment (months −3 to 0), light grey bars the dose-finding phase (months +1 and +2), dark grey bars the treatment phase on a stabile individual dose. n=28 patients (per-protocol cohort), data are depicted as median with IQR (P<0.0001). (B) Number of CELs of the SABA trial cohort plotted as orange dot in the model of the regression to the mean effect of CELs in clinical relapsing-remitting multiple sclerosis (RRMS) trials.25 The red line shows the predicted mean number of CELs at month 6 (y-axis), subject to the mean number of CELs at baseline (x-axis), the grey area comprises the 95% CI. The orange dot indicates the number actually observed in the study patients of the SABA trial (n=28). (C) Parenchymal brain volume (PBV) change comparing change during baseline phase before treatments (month −3 to month 0, M-3–0) and during treatment (months 5–8, M5–8). n=28 patients (per-protocol cohort), data are depicted individually as dots, median with IQR is indicated as bars (P=0.0081).
Primary and secondary MRI outcomes
Using a predictive model based on data from phase II and III trials in RRMS,25 this effect surpasses the reduction of CELs due to the regression to the mean effect (RTTME) below the 95% CI (see figure 2B). As predicted by the presented meta-analytic model, the RTTME would explain a reduction of the number of new CELs from a baseline mean value of 2.5 CELs in our patient cohort to 1.6 CELs at month 6, while the number was, in fact, reduced to a mean of 0.8 CELs in the ITT analysis.
Correspondingly, the number of new T2 lesions decreased significantly between baseline and treatment phase from 7.5 to 0.25 new T2 lesions per month. The effect on T2-lesion reduction was maintained throughout the extension period in those patients who chose to continue the study beyond month 8 of the trial (online supplementary figure 1). The change in T2-lesion volumes was not significantly different between baseline and treatment phase.
In addition, we observed a modest loss of brain volume during the four baseline scans (PBVC −0.12, IQR −0.36% to 0.13%) while we observed a small increase for brain volume during treatment (PBVC 0.11, IQR −0.06% to 0.6%, table 2 and figure 2C; P=0.0081).
Adverse events and safety
Two hundred and twenty adverse events (AE) were reported during the treatment phase. These were generally mild or moderate (57.7% respectively 38.6% of all AEs, table 3). The two most frequent AE entities were minor infections (n=72, 32.7%), of which the common cold accounted for about 60%, and gastrointestinal symptoms (n=38, 17.3%). The frequency of gastrointestinal AEs peaked during the first 4 weeks and decreased afterwards (online supplementary figure 2). Fifteen per cent (n=6) of the patients reported recurrent mild gastrointestinal AEs throughout the exposure time. No patient discontinued the SFE due to gastrointestinal AEs.
Adverse events and serious adverse events during baseline and treatment phases
The four serious AEs reported in our study were a fracture of the tibia after an accidental fall at month 7, an emergency minor proctologic inpatient treatment at month 20, hospital admission due to a newly diagnosed lupus erythematosus at month 25 and the fracture of both ankles after an accidental fall at month 27.
Two AEs received special attention and were intensively discussed with the data safety monitoring board and authorities. One patient developed rheumatoid arthritis at month 12, and another lupus erythematosus at month 25 (see above). After the second rheumatologic AE was reported, we reanalysed the serum samples of all patients for antinuclear antibody titres, anti-cyclic citrullinated peptide(CCP) antibodies and rheumatoid factor, which had been either not detectable or unchanged during treatment (online supplementary figure 3). Prospectively collected serum samples showed no changes in tumour necrosis factor-α levels (online supplementary table 3). It is important to note that one patient was known to have rheumatoid arthritis before entering the trial and reported no changes of her rheumatologic symptoms throughout the entire 24-month treatment phase. General laboratory monitoring throughout the study showed no significant changes in any patient.
Events during the baseline phase were documented in n=80 patients (left column) as a control for the treatment cohort (n=38, right column).
AEs during treatment phase include all patients who took at least one dose of the study drug. AEs are given as total numbers and as % in parentheses.
Clinical parameters
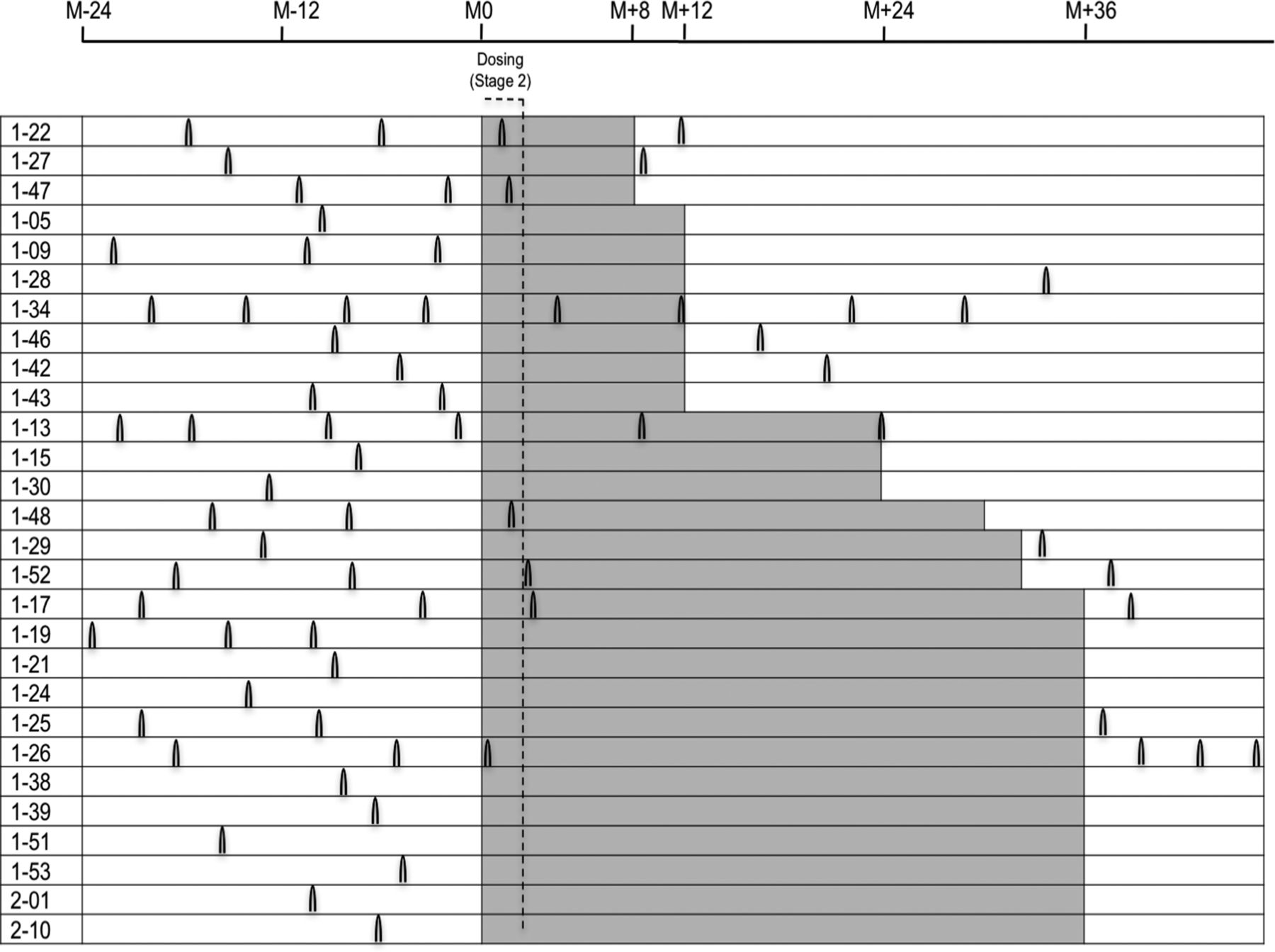
The ARR decreased from 0.93 during the year before the start of treatment to 0.48 during the first year of treatment (P=0.0422, table 4). Clinical signs of disease activity remained absent beyond month 12 in patients continuing the study (figure 3).

Occurrence of relapses in the SABA patient cohort before, during and after treatment. Relapses are indicated as black arcs; each individual patient is depicted with his/her relapses as reported 2 years before entering the trial (in white), as observed during treatment with a standardised frankincense extract (SFE; highlighted in grey) and if available during follow-up after finishing the trial (white).
Secondary and tertiary clinical outcomes
Clinical endpoints are indicated in table 4. EDSS and SNRS remained unchanged at month 8 compared with baseline. MSFC improved significantly between baseline (month 0) and months 8 and 12, respectively. SDMT improved significantly during the baseline phase, possibly due to a training effect. However, there was an additional significant improvement until month 8. PASAT improved significantly only at month 12 compared with baseline z-scores. Health-related quality improved at months 8 and 12, respectively.
Immunological parameters
Laboratory parameters as well as most lymphocyte subpopulations showed no changes throughout the first year of SFE treatment in our patient cohort (figure 4A,B online supplementary figure 4). However, we observed changes in the CD3+ T cell compartment (figure 4C–F): CD4+ T cells showed an increase in CTLA-4 surface expression, and the frequency of CD4+ CD25hi FoxP3+ T cells increased significantly during treatment. When analysing the frequency of cytokine-producing CD8+ T cells, we found that IL-17A-producing CD8+ T cells decreased while the frequency of IL-10-producing CD8+ T cells increased simultaneously during treatment (all P<0.001). Of these parameters, only the frequency of CD4+ CD25hi FoxP3+ T cells showed a significant, negative correlation with the primary outcome parameter, that is, the number of CELs (Spearman correlation coefficient −0.46, P=0.0281).

{kind=link}
{kind=link}
{kind=link}
Changes in lymphocyte subpopulations. (A, B) Leucocyte and lymphocyte count results from differential blood counts during the SABA trial (n=28 patients). Data are depicted individually as dots, median and IQR are indicated. (C, D) Regulatory CD4+ T cell markers (CTLA-4 expression, CD4+ CD25high Foxp3+ T cells). Data from n=25 patients participating in the immunological studies are depicted individually as dots, median and IQR are indicated. P values (Wilcoxon signed-rank test) comparing change from baseline (month 0 vs month 8) are as follows: CTLA-4 median fluorescence intensity (MedianFI) of CD4+ T cells: P<0.0001; CD4+ CD25hi Foxp3+ T cells in %: P<0.0001. (E, F) Cytokine-producing CD8+ T cells (interleukin (IL)-17A or IL-10-producing). Data from n=25 patients participating in the immunological studies are depicted individually as dots, median and IQR are indicated. P values (Wilcoxon signed-rank test) comparing change from baseline (month 0 vs month 8) are as follows: CD8+ IL-17+ (interferon (IFN)-gamma negative) T cells: P<0.0001; CD8+ IL-10+ (IFN-gamma negative) T cells: P=0.0005.
Using an exploratory descriptive statistics approach, we detected the following changes in the serum levels of the 14 studied cytokines (online supplementary table 3): tumour growth factor-β, IL-4 and IL-5 decreased significantly both early (months 1 and 3) and late (months 8 and 12) during the treatment phase; Granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-17A serum levels decreased significantly only during early treatment. IL-2 and IFN-γ decreased significantly only during late treatment.
Discussion
The present study is the first clinical trial of an oral anti-inflammatory treatment using an SFE in patients with RRMS. The administration of an oral SFE for 8 months was safe and well tolerated and provides safety data for the largest dose of and longest exposure time to an SFE reported in a clinical trial to date. Regarding MRI measures, the SFE significantly reduced the number and volume of CELs in this cohort of patients with RRMS. However, the sensitivity of the baseline-to-treatment trial design in regard to the RTTME poses a limitation of these observations especially in an MS patient cohort selected by high MRI disease activity. Our analysis of the ITT cohort and the use of a meta-analytic model using data from placebo cohorts of MS phase II and III trials to estimate the RTTME nevertheless indicate that the observed results exceed a sole RTTME (figure 2B).25
The reduction in the number of new T2 lesions and the increase in the PBVC evolution in MRI imply a beneficial effect of the SFE. The change of total T2 lesion volume did not differ between baseline and treatment phase, indicating near cessation of T2 lesion-causing inflammation. However, confluent lesions and oedema of recent lesions render exact measurements of the total T2 lesion volume difficult and might mask an actually existing effect. Our findings on PBVC must be discussed in the light of methodical restrictions associated with short-term atrophy measurements in MS as well as contributing factors such as lifestyle or pseudoatrophy.26 As the latter can be excluded for the four baseline assessments, pseudoatrophy might have even led to an overestimation of the true atrophy rate of the treatment phase, as an acceleration of brain volume loss was observed in other trials following the initiation of therapy with disease-modifying drugs due to the resolution of inflammation and fluid reduction.27
Immunomodulatory therapies for MS have no easily discernable, if any, effect in advanced disease stages. Consequently, it has been advocated that treatment should start soon after diagnosis, even in clinically unaffected or minimally affected patients with MS. For the latter patients there is a great and, as yet, unmet need for safe and well-tolerated oral agents to minimise the risk during long-term immunomodulatory treatment lasting two to three decades. The patients in this trial with their median EDSS of 1.5–2.0 are representative of this MS subgroup with only minor clinical impairment despite MRI evidence of rather pronounced inflammatory disease activity.
Indications of positive clinical outcomes in this trial are supporting evidence for a potential efficacy of the SFE, even though the trial was underpowered for the reliable evaluation for which phase IIb trials are performed. The reduction of the ARR from the 12 months before to the 12 months after treatment begins certainly has to be attributed in part to the RTTME and to a common form of selection bias: patients who have only recently suffered a relapse are more likely to start on immunotherapy or to enter a clinical trial. EDSS and SNRS proved to be stable throughout the observation period, while quality of life improved. The improvement in the SDMT and the PASAT results at month 12 might at least partially reflect training effects. However, a beneficial effect of the SFE on cognition in patients with RRMS cannot be excluded and was also indicated in a short-term, placebo-controlled trial using Boswellia papyrifera to treat cognitive impairment in 80 patients with MS.28
The rather high dropout rate in our trial has primarily been associated with either non-compliance to the time-consuming study protocol or with patients reconsidering approved therapies in the face of an early relapse during the follow-up. Only in two patients were side effects the reason for early withdrawal (urticaria or gustatory disgust).
Compared with approved oral MS treatments, the SFE showed a very good safety profile, particularly with respect to absolute and relative lymphocyte counts, which remained unaffected (figure 3B). Compared with the baseline observation period before the trial, the incidence of infections did not increase during treatment. Gastrointestinal AEs had been observed in previous clinical trials with SFE5 and were also the most frequent drug-related side effects in our study. Whether the two rheumatologic AEs were related to SFE or coincidental is not clear at present and should be monitored in future clinical trials.
General haematological and immunological parameters show that the treatment was not immunotoxic. While MS is a complex and heterogeneous immunological disease, there is consensus about the importance of a dysregulation of proinflammatory and anti-inflammatory T cell subsets.29 Immunological outcome data from the present trial imply a distinct immunological signature change in the T cell compartment and more specifically in the CD3+ Treg/TH17 subset, which is consistent with our in vitro observations.10 The data of this previous trial focused on CD4+ T cells, the results of the present study indicate that the immunomodulatory effects of SFE are, at least partly, due to reduced IL-17 production of CD8+ T cells. This observed effect could be related to CD8+ CD161+ TH17-like Mucosal associated invariant T (MAIT) cells that have recently received increased attention because of their possible role in the pathogenesis of RRMS.30 The fact that disease activity remained reduced for up to 36 months in patients participating in the extension phase, as evidenced by both MRI and clinical outcome parameters, supports the conclusion that SFE has a durable effect as long as treatment continues (figure 3).
In summary, the MRI, clinical and immunological data in this SABA phase IIa trial indicate that this SFE is safe and shows beneficial immunomodulatory effects in RRMS, which should be further investigated in randomised controlled phase IIb or III trials. The combined analysis of MRI, clinical and immunological outcomes in an open baseline-to-treatment design with frequent monitoring is attractive for patients and investigators for early proof-of-concept studies to evaluate new treatment approaches in small patient groups. Furthermore, due to its open design and frequent monitoring, it is also attractive from a safety perspective since investigator and patient are aware of the disease activity and can discuss a switch to a different treatment if unexpected safety issues or increased disease activity should occur. Daclizumab (anti-CD25) can be referred to as one example. Here, the effects on activity seen in MRI and the reduction of ARR that had been observed in an even smaller trial with a similar design31 endured throughout clinical development and large-scale phase III testing.32 However, despite our encouraging results, it is difficult to forecast the efficacy of an SFE in RRMS, and larger trials are required to demonstrate an effect on clinical outcome. It is also clear that, despite a growing list of treatment options, the right balance between efficacy and safety profile becomes increasingly more important for a young patient population, who require long-term treatment. The results of the SABA trial are promising from this perspective.
Acknowledgments
We thank all our patients who participated in this trial. We thank Dan Nguyen Luong for technical assistance with the Mesoscale measurements. We thank Thomas Crozier for editorial assistance for non-intellectual content. We thank Alpinia Laudanum for providing us with a standardised frankincense extract free of charge.
References
Footnotes
Data Safety Monitoring Board Alessandra Solari (S.O.S.D. Neuroepidemiologia, Fondazione IRCCS Istituto Neurologico “C. Besta”, Milano, Italy), Ingo Kleiter (Neurologische Klinik, Ruhr Universität Bochum, Germany), Brigitte Wildemann (Neurologische Klinik, Universität Heidelberg, Heidelberg, Germany).
Contributors KHS, CH and RM designed the study. KHS, JPS, CH, JD, FP, S Schammler, SR, SF, LAIV, FH and OP collected the data. KHS, JPS, CH, JD, FP, SFl, S Siemonsen, GW, TF, OW, LAIV, FH and OP analysed the data. KHS and TF performed the statistical analysis. KHS, CH, OP and RM wrote the report. KHS, JPS, JD, FP, SF, S Schammler, SR, S Siemonsen, GW, TDF, TF, OW, LAIV and FH revised the report. All authors had full access to all of the data in the study. The corresponding author had the final responsibility for the decision to submit for publication.
Funding This work was supported by the Bundesministerium für Bildung und Forschung (BMBF, Förderkennzeichen 0315610-0315620 and Förderkennzeichen 16GW0082) in the context of NEU2. The INIMS at the Universitätsklinikum Hamburg-Eppendorf has been supported by the Hertie-Stiftung. The extension study was supported by Alpinia Laudanum, Switzerland. None of the funding sources had any role in designing the study, in collection, analysis, or interpretation of data, or in writing the article.
Competing interests KHS, JPS and CH report grants from the Bundesministerium für Bildung und Forschung, grants and provision of study drug from Alpinia Laudanum, during the conduct of the study. KHS reports grants from Biogen, outside the submitted work. JPS reports grants and personal fees from Biogen, personal fees from Genzyme, grants and personal fees from Novartis, grants from Merck Serono, outside the submitted work. JD reports grants, personal fees and non-financial support from Bayer Healthcare; grants, personal fees and non-financial support from Novartis, personal fees and non-financial support from Biogen, personal fees from Sanofi-Genzyme, personal fees from Allergan, personal fees from Merck Serono, outside the submitted work. FP reports grants and personal fees from various pharmaceutical companies, outside the submitted work. TF reports personal fees from Novartis, personal fees from Bayer, personal fees from Biogen, personal fees from AstraZeneca, personal fees from Janssen, personal fees from Grünenthal, personal fees from Pharmalog, personal fees from SGS, personal fees from Roche, outside the submitted work. OP and LAIV report grants from Grant from the German Federal Ministry of Education and Research-BMBF (16GW0082), during the conduct of the study. CH has received funding for projects and invited talks from the following pharmaceutical companies: Biogen, Genzyme, Merck Serono, Roche. RM reports grants from the Ministry of Education and Research, grants from European Research Council, grants from Swiss National Science Foundation, grants from Swiss Commission for Technology and Innovation, grants from German Research Society, grants from Swiss MS Society, during the conduct of the study, grants and personal fees from Biogen, personal fees from Genzyme Sanofi Aventis, grants and personal fees from Novartis, personal fees from Roche, personal fees from Teva, personal fees from Merck AG, grants and personal fees from Cellprotect, personal fees from Neuway Pharma, outside the submitted work. GW, S Siemonsen, SR, SF, FH, TDF, OW and S Schammler declare no competing interest.
Ethics approval German Federal Institute for Drugs and Medical Devices (BfArM, Approval No. 4036771) and the local ethics committees (Approval No. PVN3389 and ZS EK 13572/09).
Provenance and peer review Not commissioned; externally peer reviewed.