
Abstract
The neuronal cytoskeleton is tightly regulated by phosphorylation and dephosphorylation reactions mediated by numerous associated kinases, phosphatases and their regulators. Defects in the relative kinase and phosphatase activities and/or deregulation of compartment-specific phosphorylation result in neurodegenerative disorders. The largest family of cytoskeletal proteins in mammalian cells is the superfamily of intermediate filaments (IFs). The neurofilament (NF) proteins are the major IFs. Aggregated forms of hyperphosphorylated tau and phosphorylated NFs are found in pathological cell body accumulations in the central nervous system of patients suffering from Alzheimer’s disease, Parkinson’s disease, and Amyotrophic Lateral Sclerosis. The precise mechanisms for this compartment-specific phosphorylation of cytoskeletal proteins are not completely understood. In this review, we focus on the mechanisms of neurofilament phosphorylation in normal physiology and neurodegenerative diseases. We also address the recent breakthroughs in our understanding the role of different kinases and phosphatases involved in regulating the phosphorylation status of the NFs. In addition, special emphasis has been given to describe the role of phosphatases and Pin1 in phosphorylation of NFs.
Similar content being viewed by others

Introduction
Neurons are highly specialized in the transmission and processing of electrical and chemical signals, and have unique structural characteristics. With axons in the peripheral nervous system (PNS) several meters in length, it is not surprising that a stable but dynamic structure is essential to maintain the shape of these cells, which is provided by a neuron-specific cytoskeleton. The largest family of cytoskeletal proteins in mammalian cells is the superfamily of the IFs of which the NF proteins are the neuron-specific IFs. NFs are classified as type IV IFs and classically comprise three subunits of different molecular masses: neurofilament light (NFL; 68 kDa), neurofilament medium (NFM; 145–160 kDa), and neurofilament heavy (NFH; 160–200 kDa) (Sihag et al. 2007). Later, α-internexin, another type IV IF expressed in the central nervous system, was considered as a fourth subunit (Yuan et al. 2006), while peripherin, a type III IF expressed in the PNS, is able to interact with the NFs (Beaulieu et al. 1999). The NFs are the most abundant elements of the adult axonal cytoskeleton, which underscore their importance in neuronal functions. Their most essential function is regulating axonal diameter and thereby increasing conduction velocity (Friede and Samorajski 1970; Zhu et al. 1997). Furthermore, they are involved in neuronal differentiation, axon outgrowth, and regeneration. By participating in these active processes, it is now clear that NFs are more dynamic than initially postulated (Angelides et al. 1989; Shea et al. 2009).
All NFs comprise a central helical coiled-coil domain flanked by a globular amino-terminal head domain and a carboxyl-terminal tail domain. NF-H and NF-M, contain hypervariable Lys/Ser/Proline (KSP) repeats in carboxyl-terminal tail domain that is absent in NF-L (Xu et al. 1994; Nixon and Shea 1992; Lee and Cleveland 1996). The central rod domain is involved in the coiled-coil formation of the filamentous structure, while the globular amino-terminal head domain is involved in NF assembly. The carboxyl-terminal domain forms sidearms that extend from the filament and appear to form connections between adjacent NFs and adjacent structures. The most striking feature of the sidearms in NF-H and NF-M is the lysine/serine/proline (KSP) repeat motifs that are phosphorylated by cyclin-dependent kinase-5 (Cdk5) and other proline-directed kinases. These repeats are extensively phosphorylated in axons in vivo, and due to these motifs, NF-H is one of the most extensively phosphorylated neuronal proteins. Mass spectroscopic analyses of NF-H in humans, rats, canines, and squid have shown that most of the KSP repeats are phosphorylated in vivo (Bennett et al. 1981; Shetty et al. 1993; Jaffe et al. 2001).
Neurofilament Proteins (NF-M/H) are extensively phosphorylated in AD brains
The relative contributions of hyperphosphorylated tau and NFPs to the tangle pathology seen in AD and ALS are uncertain and controversial. Phosphorylated epitopes of NF-M/H have been reported in tangles utilizing specific monoclonal antibodies (Sternberger et al. 1985). However, these methods do not precisely determine the number and specific phosphorylated residues in a given protein nor do they distinguish between the phosphorylation of specific S/T residues in KSP or KTP repeats or in other motifs. We utilized iTRAQ and pulsed Q dissociation (PQD) methodology to characterize and quantify phosphorylation sites of NF-M and NF-H in AD brain. We identified 13 phosphorylated Ser/Thr sites of NF-M that are phosphorylated at fourfold to eightfold greater abundance in AD brain compared with control brain. Ten phosphorylated KSP sites have been identified on the C-terminal tail domain of NF-H, with greater abundance of phosphorylation in AD brain compared with control brain. These findings of a significant number of phosphorylation sites in NF-M and NF-H that are proline-directed Ser/Thr (S/T-P) residues suggests that either proline-directed kinases [Cdk5, glycogen synthase kinase 3 beta (GSK3β) or mitogen-activated protein kinases (MAPKs)] are hyperactivated or the protein phosphatases [e.g., protein phosphatase 2A (PP2A)] are down-regulated. Furthermore, identification of non-SP phosphorylation sites in NF-M (Ser-783 and Ser-788) at a significantly higher level in AD suggests the involvement of non-proline-directed phosphorylation of NF-M/H in AD brain. This study represents the first comprehensive iTRAQ analysis and quantification of phosphorylation sites of human NF-M and NF-H from AD brain and confirms that aberrant hyperphosphorylation of neuronal intermediate filament proteins is involved in AD (Rudrabhatla et al. 2010). The study has been extended to an iTRAQ analysis of isolated preparations of purified neurofibrillary tangles (NFT) from AD brains, and we reported direct evidence on the identity of NFs in NFTs. In addition to several taw phosphatase the phosphoproteomics of NFTs clearly identified NF-M and NF-H phosphopeptides. The proteomics analysis also identified an NF-L peptide and another intermediate filament protein, vimentin. Finally, phosphopeptides corresponding to microtubule-associated protein 1B (MAP1B) and microtubule-associated protein 2 (MAP2) were also identified. In corresponding matched control preparations of NFTs, none of these phosphorylated neuronal cytoskeletal proteins were found. These studies independently demonstrate that NF proteins are an integral part of NFTs in AD brains (Rudrabhatla et al. 2011).
Compartment-specific regulation of neurofilament phosphorylation
Neuronal topographic regulation refers to compartment-specific activities of kinases and phosphatases regulating the phosphorylation of neuronal cytoskeletal elements, in health and disease. For example, the numerous KSP phosphorylation sites in NF-M and NF-H C-terminal tail domains are primarily and stably phosphorylated in axons, whereas N-terminal head domain sites are transiently phosphorylated in cell bodies (Rudrabhatla and Pant 2010; Nixon 1993; Nixon et al. 1994; Zheng et al. 2003; Grant et al. 2006; Sihag et al. 2007). This topographic pattern of phosphorylation is tightly regulated in the nervous system. Deregulation of this post-translational modification has been implicated in various neurodegenerative diseases. The phosphorylation of KSP motif-rich tail domains on NF-M and NF-H might be regulated by the activation of proline-directed kinases [extracellular-signal-regulated kinases 1/2 (ERK1/2), Cdk5, p38 MAP kinase, or stress-activated protein kinases (SAPK)]. Cdk5 is one of the principal kinases involved, together with Erk1/2 and SAPK kinases (Giasson and Mushynski 1997; Veeranna et al. 2000). Deregulation of these kinases, such as hyperactivation of Cdk5 due to neuronal insults (e.g., oxidative stress, Aβ toxicity, glutamate toxicity), leads to neurodegenerative pathology, intraperikaryal accumulation of hyperphosphorylated cytoskeletal proteins, tangles of phospho-tau and aggregated phosphorylated neurofilaments and neuronal death, hallmark pathologies of AD, ALS, and dementia with Lewy bodies (Patrick et al. 1999; Kesavapany et al. 2004).
Aggregation of phospho-NFs is a hallmark in various neurodegenerative diseases such as AD, ALS, PD, diabetic neuropathy, and giant axonal neuropathy (Liu et al. 2011). The phospho-NF aggregates are found in the cell body and proximal parts of the axons, whereas normally phospho-NFs only reside in the distal parts of the axons (Shea et al. 2009). Such abnormal phosphorylated NF aggregates are also observed in transgenic mutant NFL and wild-type NFH overexpressing mouse models, suggesting that changes in the stoichiometry of the NFs are a critical point to consider (Cote et al. 1993; Xu et al. 1993; Liu et al. 2011). Mice overexpressing NFH show specific degeneration of large motor neurons in the spinal cord resembling the situation in ALS (Cote et al. 1993; Lee et al. 1994). Abnormal phosphorylation of NFs can inhibit their proteolytic breakdown (Goldstein et al. 1987; Pant 1988). The phospho-NF aggregates may become cytotoxic and interact inappropriately with other cytoskeletal elements of the perikarya (Zhou et al. 2010). The increased time they spend in the cell body is thought to result in further aberrant phosphorylation (Black and Lee 1988) and may prevent them from entering the axon, resulting in a deleterious feedback loop (Sihag et al. 2007). Moreover, kinesin and dynein motor proteins were found to accumulate in these NF spheroids (Toyoshima et al. 1998), resulting in motor entrapment and eventually impairment of the overall axonal transport (Collard et al. 1995). Various stress factors [osmotic, oxidative, UV, glutamate, amyloid beta (Aβ) amyloid] were shown to have the ability to activate a kinase involved in NF phosphorylation. For instance, MAPK kinase kinase (MEKK1) is known to mediate JNK activation in response to stress but depending on the insult MEKK1 can also activate ERK (Giasson and Mushynski 1997).
Phosphorylation has long been considered to regulate NF interaction and axonal transport, and, in turn, to influence axonal stability and maturation to large-caliber axons. Cdk5, a serine/threonine kinase homologous to the mitotic cyclin-dependent kinases, phosphorylates NF subunits in intact cells. Previously, we demonstrate that overexpression of Cdk5 increases NF phosphorylation and inhibits NF axonal transport, whereas specific inhibition of cdk5 reduces NF phosphorylation and enhances NF axonal transport in cultured chicken dorsal-root-ganglion neurons (Shea et al. 2004a, b). Large phosphorylated NF “bundles” were prominent in perikarya following Cdk5 overexpression. These findings suggest that Cdk5-p35 activity regulates normal NF distribution and that overexpression of Cdk5-p35 induces perikaryal accumulation of phosphorylated NFs similar to those observed under pathological conditions (Shea et al. 2004a, b).
In the motor neurons of mutant ALS mouse models and ALS patients, active p38 was found in the phospho-NF aggregates (Tortarolo et al. 2003; Ackerley et al. 2004). Furthermore, an increase in reactive oxygen species (ROS) production was shown to activate Cdk5, resulting in perikaryal NF deposits and slower NF axonal transport in cortical neurons. Neuronal stress is a factor in several neurodegenerative diseases such as AD, ALS, PD, and Huntington’s disease (HD), where Cdk5 is shown to be overactive (Shea et al. 2004a). Any kind of stress leading to an intracellular increase in Ca2+ can cause calpain over activity. This subsequently results in the cleavage of the neuronal Cdk5 activator p35 to its 25 kDa proteolytic fragment (p25) (Patrick et al. 1999). The latter has a longer half-life and is not readily degraded, and by binding to Cdk5, it changes the cellular location and substrate specificity of this kinase. As NF proteins are major Cdk5 substrates, it has been suggested that they may act as a phosphorylation sink for deregulated Cdk5 activity in ALS (Nguyen et al. 2001), resulting in NF hyperphosphorylation (Zhou et al. 2010). Interestingly, NF abnormalities can be an early marker of pathogenesis (Xiao et al. 2006) as observed in the ALS mutant SOD1 transgenic mouse where the slowing of axonal transport and NF accumulation in the cell body is observed before any loss of motor function. While the cell is degenerating, cell type-specific proteins such as phospho-NFs are released into the extracellular matrix (ECM). The ECM is in equilibrium with the cerebrospinal fluid (CSF), and in the latter, the neuron-specific proteins can be detected. Therefore, phosphor NFs are used as early protein biomarkers in many neurodegenerative disorders (Petzold 2005; Brettschneider et al. 2006a, b), as they can be detected in the CSF before any clinical symptoms occur. In conclusion, although many molecular aspects of NF dynamics and transport are still unclear or controversial, there is increasing evidence that phosphorylation is pivotal for the proper functioning of NFs and other cytoskeletal proteins. Further effort is necessary in the dissection of molecular pathways underlying NF phosphorylation.
Protein phosphatases (PPs) and NF phosphorylation
The protein kinases that phosphorylate NF, amyloid precursor protein (APP), tau, and the proteases involved in generation of amyloid beta (Aβ) are clearly important in studies of neurodegeneration. However, it has also been recognized that the protein phosphatases that reverse the actions of these protein kinases are equally important and warrant detailed analysis. Protein phosphatases (PPs) are classified on the basis of their ability to dephosphorylate either serine and threonine residues or tyrosine residues (protein tyrosine phosphatase, PTPs, or dual-specificity DSPs). The PPs family includes PP1, PP2A, PP2B (also known as calcineurin), PP4, PP5, and PP6. High levels of the serine/threonine PPs are found throughout the brain, with PP1, PP2A, PP2B, and PP5 being abundant and implicated in AD. Recent studies have also begun to reveal roles for PTPs, especially striatal-enriched tyrosine phosphatase, in AD (Böhmer et al. 2013). Over the past decade, interest in the role of phosphatases has been intensified (Pei et al. 1997, 1998; Liu et al. 2005a). However, but much less is known about the role of PPs in regulating cytoskeletal protein phosphorylation, especially those acting on proline-directed Ser/Thr phosphorylation. The reported turnover of phosphate groups on NFs during axonal transport and the decrease in number of phosphorylated epitopes of NF proteins at the nodes of Ranvier relative to the internodal myelinated regions demonstrate the physiological significance of PPs in regulating the state of NF phosphorylation. More significantly, phosphatases have been implicated in the regulation of cytoskeletal tau phosphorylation in AD (Liu et al. 2005a, b). AD brains contain reduced levels of PP2A and other phosphatases (Vogelsberg-Ragaglia et al. 2001). Tau phosphorylation is regulated in AD brains by PP2A acting at a specific residue, Thr 236. It is becoming increasingly clear that some of the pathology seen in neurodegenerative disorders may be attributed to defects in the regulation of the synthesis and activities of several key neuronal phosphatases.
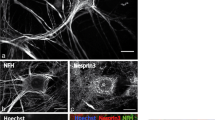
Previous studies from our laboratory have reported that PP2A from both rat spinal cord and rabbit skeletal muscle can dephosphorylate the NF-M/H tail domain following phosphorylation by Cdk5 (Veeranna et al. 1995). Aberrant hyperphosphorylation of NF-M/H causes NFs to form aggregates of phospho-NF-M/H in neuronal perikarya in AD and ALS. Figure 1A shows the schematic representation of NF-M/H and their KSP repeats in their tail domains which are selectively phosphorylated in the axonal compartment under physiological conditions. Our understanding of this phenomenon has improved over the past decade, although the mechanism underlying the aberrant hyperphosphorylation of NF under pathological conditions is still unclear (Veeranna et al. 1998; Grant and Pant 2000; Zheng et al. 2003; Sihag et al. 2007). Hyperactivity of proline-directed kinases, such as Cdk5, MAPKs, and GSK3β, have been shown to be implicated in the aberrant phosphorylation of NF, but the role of phosphatases has not yet been explored. Compared with the hundreds of known kinases, there are only a handful of phosphatases, and each must dephosphorylate a wide range of protein substrates. Previous studies have suggested that Ser/Thr phosphatases are involved in neurodegeneration (Vogelsberg-Ragaglia et al. 2001). We found that the PP2A activity is reduced in ALS spinal cord and AD brain (Kesavapany et al. 2007). In a recent study, we have used phosphatase inhibitors to study NF phosphorylation in cortical neurons (Rudrabhatla et al. 2009). The data presented in Fig. 1B, a–d demonstrate that PP2A-specific inhibitors, such as okadaic acid (OA), microcystin LR, and fostriecin, cause aberrant hyperphosphorylation of NF-M/H in perikarya of cortical neurons in vitro, resembling that found in AD and ALS. Conversely, the PP2B-specific inhibitor cyclosporine A did not induce perikaryal aberrant hyperphosphorylation of NF even at high concentration (Fig. 1B–E) (Rudrabhatla et al. 2009). We and other laboratories have shown that PP2A is the major NF and tau phosphatase, while PP2B contributes less toward the dephosphorylation of NF. Consistent with this observation, Strack et al. (1997) reported that PP2A and PP1 were associated with the spinal cord NF fraction, but PP2B was found exclusively in the low molecular weight fraction. They attributed 75 % of NF dephosphorylation to PP2A and the rest to PP1. Quantitation of PP2A and PP2B in perikarya is shown in Fig. 1C. The data presented in Fig. 1D demonstrate that PP2A colocalizes with phospho-NFs in OA treated cortical neurons. We showed that cyclosporine A which inhibits PP2B had no effect on perikaryal phosphorylation of NF (Rudrabhatla et al. 2009 ). It has recently been confirmed that phosphorylated NF-H increases in the rat CNS during aging (Gou and Leterrier 1995) consistent with a significant decline in phosphatase activities, particularly PP2A. (Veeranna et al. 2011). These studies suggest that PP2A may be major phosphatase regulating topographic NF phosphorylation and is a target for therapeutic intervention in those neurodegenerative disorders exhibiting a decline in PP2A activity, as in AD.

PP2A is the major phosphatase regulating topographic phosphorylation of neurofilaments. A Schematic of NF-M/H showing KSP repeats in the tail domain. The KSP repeats are recognized by phospho-NF-M/H antibodies (SMI31, SMI34) and phospho-NF-H antibodies (RT97). B Dissociated E18 rat cortical neurons were cultured for 7 days in culture (7DIC) and then treated with or without different protein phosphatase PP2A-specific inhibitors for 2.5 h [OA (b 0.25 μM), microcystin LR (c 0.5 μM), or Fos (d 1.05 μM)] and then subjected to ICC assay with phospho-NF (SMI31) antibodies. Control (a) shows phosphorylated NF in processes with no phosphorylation in cell bodies. There was a significant increase in somatic phosphorylation in OA-treated (b), mLR-treated (c), and fostriecin-treated neurons (d). (e) Neurons treated with the PP2B-specific inhibitor (20 μM) show no effect on NF phosphorylation. C Densitometric analyses of p-NF were performed with images captured using NIH Image software. Similar results were obtained using RT97 and SMI34 antibodies (**p < 0.05; *p < 0.1). D PP2A colocalizes with phospho-NF in neuronal perikarya in OA-treated cortical neurons. PP2A (green), phospho-NFs (red), DAPI (blue), yellow overlap between PP2A and phospho-NFs. E Pin1 modulates OA-induced perikaryal phosphorylation. Rat cortical neurons (5 DIC) were transfected with either control (Ctrl) and scrambled siRNA or Pin1 siRNA and then treated with 0.25 μM OA on day 7 for 2.5 h. Neurons were immunostained; p-NF-H was detected using SMI31 (red), and Pin1 was detected with Pin1 antibody (green). Neurons transfected with Pin1 siRNA exhibited reduced p-NF-H in the cell body. Non-treated neurons exhibited SMI31 staining in the processes with little or no staining in the cell body (a–c), which increased during OA treatment (d–f). Cell body SMI31 staining was reduced in Pin1 siRNA-treated neurons (g–i). Arrows point to cell bodies
Peptidyl-prolyl cis/trans isomerase 1 (Pin1) and neurofilament phosphorylation
Pin1 is a peptidyl-prolyl isomerase originally identified in a genetic screen for proteins involved in mitotic regulation and was found to be essential for cell division in some organisms. It specifically isomerizes phosphorylated (Ser/Thr)-Pro motifs and thus plays a post-phosphorylation role in regulating protein function. Structural and functional analysis of Pin1 suggests that substrate recognition is phosphorylation dependent. Pin1 is localized in both nuclei and cytoplasm of most cells. Pin1-dependent prolyl isomerization promotes PP2A-mediated dephosphorylation of certain phosphorylated Ser/Thr-Pro motifs. Within the last decade, a number of studies have identified Pin1 as a key player in regulating phosphorylation of several mitotic proteins during mitosis and cytoskeletal proteins in neurons during development and neurodegeneration (Driver and Lu 2010; Lu et al. 2003; Ramakrishnan et al. 2003; Hamdane et al. 2006). Pin1 targets phosphorylated Ser-Proline and Thr-Proline residues switching cis to trans configurations. This, in addition to phosphorylation itself, acts as a conformational switch that changes activity and function of the target protein. Pin1 contains an N-terminal WW domain and C-terminal isomerase domain. The WW domain functions as a specific phospho-Ser/Thr binding module, interacting with specific phospho-Ser/Thr-Pro motifs present in a defined subset of phosphoprotein substrates; it induces cis/trans isomerization leading to conformational changes and specific function of the target protein. Its role in regulating tau phosphorylation in AD has been particularly highlighted with suggestions that one specific site Thr-231 phosphorylation is modulated by Pin1 and, hence, may play a role in regulating tau phosphorylation in neurodegeneration (Landrieu et al. 2011).
In recent years, our interest in Pin1 is based on the fact that neurofilaments, the most abundant neuronal proteins which contain many phospho-acceptor sites (KS/TP) in tandem, are also aberrantly hyperphosphorylated in AD and ALS neurons (Kesavapany et al. 2007). We have shown that Pin1 associates with phosphorylated neurofilament-H (p-NF-H) in neurons and is colocalized in ALS-affected spinal cord neuronal inclusions. To mimic the pathology of neurodegeneration, we demonstrated that glutamate-stressed neurons displayed increased p-NF-H in perikaryal accumulations that colocalized with Pin1 and led to cell death. Both effects were reduced upon inhibition of Pin1 activity by the use of an inhibitor juglone and down-regulating Pin1 levels through the use of Pin1 small interfering RNA (Si RNA). Thus, isomerization of Lys-Ser-Pro repeat residues that are abundant in NF-H tail domains by Pin1 can regulate NF-H phosphorylation. In an in vitro kinase assay, the addition of Pin1 substantially increased phosphorylation of NF-H KSP repeats by proline-directed kinases, Erk1/2, Cdk5/p35, and c-Jun N-terminal protein kinases (JNKs) in a concentration-dependent manner. Because oxidative stress plays an important role in the pathogenesis of neurodegenerative diseases, we studied the role of Pin1 in stressed cortical neurons and human embryonic kidney 293 (HEK293) cells. Both H2O2 and heat stress induce phosphorylation of NF-H in transfected HEK293 cells and primary cortical cultures resulting in perikaryal phospho-NF-H accumulations, and neuronal apoptosis. Knockdown of Pin1 by transfected Pin1 short interference RNA and DN-Pin1 rescues the effect of stress-induced NF-H phosphorylation. JNK3, a brain-specific JNK isoform, is activated under oxidative and heat stress, and inhibition of Pin1 by Pin1 short interference RNA and DN Pin1 inhibits this pathway (Kesavapany et al. 2007; Rudrabhatla et al. 2009). Our results implicate Pin1 as a possible modulator of stress-induced NF-H phosphorylation as seen in neurodegenerative disorders like AD and ALS. Thus, Pin1 may be a potential therapeutic target for these diseases.
Indeed, Pin 1 is implicated in neurodegeneration Pin1 also localizes to the Lewy bodies (LBs), in PD brain tissue and thereby enhances the formation of α-synuclein immunoreactive inclusions (Ryo et al. 2006). In a cellular model of α-synuclein aggregation, Pin1 overexpression facilitates the formation of α-synuclein inclusions, whereas inhibition of Pin1 by dominant-negative Pin1 expression suppresses this process. Pin1 binds synphilin-1 and enhances its interaction with α-synuclein, thus likely facilitating the formation of α-synuclein inclusions. Pin1-dependent prolyl isomerization plays an important role in a post-translational modification pathway for α-synuclein aggregation and results in Lewy body formations in PD (Ryo et al. 2006). Reduction in Pin1 or its inhibition in AD seems to be correlated with tau hyperphosphorylation and tangle formation, which triggers abnormal neuronal amyloid deposition and cell death (Holzer et al. 2002; Lu et al. 2003; Butterfield et al. 2006; Pastorino et al. 2006). In AD brain tissue, Pin1 accumulates in pathological neurofibrillary tangles. By catalyzing the cis/trans isomerization and dephosphorylation of phospho-Thr 231-p-Tau by PP2A, Pin1 restores the ability of phosphorylated tau to dissociate from microtubules (Lu et al. 1999). Finally, Pin1-knockout mice express progressive age-dependent neurodegeneration, which is characterized by tau hyperphosphorylation, motor, and behavioral deficits (Liou et al. 2002).
The question arises as to the mechanism of Pin 1 action. In vitro, the addition of Pin1 actively promotes the phosphorylation and stabilization of NF-M/H by proline-directed kinases (Rudrabhatla et al. 2009). It is suggested that Pin1 normally regulates axonal NF phosphorylation of C-terminal KSP sites, stabilizing the NF polymer by cis–trans isomerization. As to its role in neurodegeneration under stress, the perikaryal proline-directed kinases are hyperactivated and phosphorylate cytoskeletal proteins; Pin1, in turn, induces cis/trans isomerization, stabilizes this phosphorylation and aggregation of phospho-NFPs (Rudrabhatla et al. 2009).
Pin1 also modulates phosphatase activity via its regulation of cis–trans isomerization of phosphorylated Ser/Thr-prolyl residues in tau and NF proteins. We have shown that exposure of cortical neurons to OA induces a dramatic increase in perikaryal NF phosphorylation, presumably as a result of inhibition of the phosphatase PP2A (Rudrabhatla et al. 2009). Data presented in Fig. 1E show that Pin1 modulates the OA-induced perikaryal phosphorylation in cortical neurons. Inhibition of Pin1 by Pin1 siRNA or DN Pin1 reduced p-NF-M/H in perikarya. The PP2A inhibition induces aberrant hyperphosphorylation of NFPs and cells undergo apoptosis. Pin1 modulates this process since inhibition of Pin1 by siRNA and/or DNPin1 inhibited the OA effect; the pathology is eliminated and cells are rescued (Fig. 1E, a–i). Inhibition of PP2A also disrupted NF axonal transport, which was rescued upon inhibition of Pin1 (see Fig. 2A, a–i). Regulation of NF phosphorylation by Pin1 differs from its effect on tau hyperphosphorylation. In the latter case, Pin1 targets a specific phosphorylated residue, Thr 231, inducing a cis–trans change that invites PP2A dephosphorylation restoring tau function (Lu et al. 1999). Pin1 interaction with phosphorylated NF-M/H with its numerous KSP C-terminal repeats evokes different biological effects and functions.

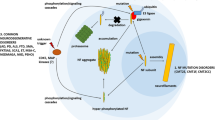
A Pin1 modulates NF transport of OA-treated neurons. GFP-labeled NFH was transfected into 5 DIC cortical neurons. After 24 h translocated GFP–NFH was monitored in cells by immunoassay and shown to colocalize in neurites with TuJ1 (a–c) suggesting NFH transport from the cell body. Treatment with OA for 2 h, however, induces perikaryal hyperphosphorylation of NFH which inhibits NF transport to distal end of neurites (d–f). Knockdown of Pin1 with Pin1 SiRNA rescues NFH transport into neurites in OA-treated neurons (note extensive overlap of GFP–NFH and TuJ1 staining in neurites, g–i). B–D Proposed model: Pin1 and regulation of topographic phosphorylation of NF B. a Schematic representation of triplet NF subunits. NF proteins form dimers/tetramers upon their synthesis in the cell bodies. b NF head domains are transiently phosphorylated by second messenger kinases. The transient phosphorylation prevents assembly into polymers and also inhibits the phosphorylation of KSP repeats in the tail domain. Tail domain unphosphorylated KSP domains remain in a compact globular form (blue circles) in the cell bodies. Although Pin1 is expressed in both cell body and axonal compartments, it fails to act on non-phospho-proline-directed Ser/Thr residues. C NF-dimers/tetramers move to the axon hillock region, where the N terminus is dephosphorylated by high levels of PP2A. NF dimers/tetramers assemble into nascent 10 nm NFs and NF transport begins. In the axon, proline-directed kinases, activated by glial/axonal interaction, phosphorylate a few tail domain KSP motifs (seen schematically below). Phosphorylated tail domains extend into side chains interacting with one another and with microtubules, forming an axonal cytoskeletal lattice. D Pin1 plays a key role in modulating the dynamics of this process in the axon. The cartoon shows the first three adjacent KSP repeat units (SP1–SP2) in the tail domain of NFH. Phosphorylation of the first two SP1 and SP2 by proline-directed kinase is shown. Using the SP2 as an example, phosphorylation induces a spontaneous conformational change from a trans to a cis configuration and blocks additional phosphorylation in the absence of Pin1. However, Pin1 induces a cis–trans isomerization, stabilizes this phosphorylation, and increases the accessibility of the proline-directed kinase and phosphorylation of the next KSP residue, SP3. Sequential phosphorylation and Pin1 stabilization of all KSP residues continue along the C-terminal domain as it unfolds into a sidearm. On the other hand, Pin1 induced cis/trans isomerization of proline-directed Ser/Thr residues of NF-M/H in the perikarya of stressed neurons, and premature extension of NF-M/H sidearms may prevent NF transport out of perikarya, causing aggregation of pNFPs and pathology
On the basis of our recent studies, we have proposed a model to explain the role of Pin1 in stabilizing the NF-M/H phosphorylation in health and disease. The model is presented in Fig. 2B–D. It describes the biogenesis of NFPs, their assembly, transport, phosphorylation, and stabilization in the axonal compartment under normal conditions. (See legends to Fig. 2 for details of Pin1 modulation of NF processing). Neuronal insults due to mutations and environmental factors cause aberrant hyperphosphorylation of cytoskeletal proteins and stabilization by Pin1 resulting in pathology. This newly defined role of Pin1 in modulating kinase and phosphatase activities and stabilizing the compartment-specific Ser/Thr-Pro phosphorylation in neurons in physiology and pathology suggests that Pin1 may be an excellent candidate for therapeutic studies in AD and ALS.
Discussion
Neurofilaments (NFs) are the neuron-specific IFs that participate in maintaining the cytoskeletal architecture of neurons. NFs are extensively phosphorylated, and the activation of the proline-directed kinases Cdk5 and ERK1/2 is primarily responsible for the extensive axonal phosphorylation of multiple KSP repeats within the carboxyl-terminal domains of NF-M and NF-H. The phosphorylation of NFs is tightly regulated, and Cdk5 activity regulates ERK1/2 activity and its NF phosphorylation. Understanding the molecular mechanisms by which the activities of kinases and phosphatases are modulated in neurodegenerative diseases can reveal novel insights into the role of aberrant phosphorylation in pathogenesis. We have described how aberrant phosphorylation of the NFs may play in neurodegenerative diseases. Transgenic mouse models have also been helpful in delineating the role of hyperphosphorylation of NFs in various neurodegenerative diseases. We have shown that hyperphosphorylation of proline directed kinases such as Cdk5 contribute to cytoskeletal pathology resembling that seen in AD (Rudrabhatla et al. 2009; Sundaram et al. 2013). In the course of studies of Cdk5 regulation, we have discovered a 127 aa fragment of the Cdk5 activator p35, CIP, is a potent inhibitor of p25-mediated Cdk5 hyperactivity without affecting “normal” p35-mediated Cdk5 activity in neurons. CIP selectively inhibits p25/Cdk5 hyperactivity and suppress the aberrant tau phosphorylation in cortical neurons (Zheng et al. 2005). In a proof of concepts study CIP peptide inhibits abnormal p25/Cdk5 hyperactivity in a p25 expressing mouse model, reduces the neurofilament hyperphosphorylation, and significantly rescues AD pathology (Sundaram et al. 2013). In addition, further insight into interactions between NFs, phosphatases and Pin1, link cytoskeletal elements together and connect them to various neurodegenerative diseases may help us to unravell the complete signaling pathways regulating phosphorylation and hyperphosphorylation of NFs and including the role of Pin1. These studies might help us to identify targeted therapies, particularly for neurodegenerative diseases.
References
Ackerley S, Grierson AJ, Banner S, Perkinton MS, Brownlees J, Byers HL, Ward M, Thornhill P, Hussain K, Waby JS, Anderton BH, Cooper JD, Dingwall C, Leigh PN, Shaw CE, Miller CC (2004) p38alpha stress-activated protein kinase phosphorylates neurofilaments and is associated with neurofilament pathology in amyotrophic lateral sclerosis. Mol Cell Neurosci 26:354–364
Angelides KJ, Smith KE, Takeda M (1989) Assembly and exchange of intermediate filament proteins of neurons: neurofilaments are dynamic structures. J Cell Biol 108:1495–1506
Bajaj NP (2000) Cyclin-dependent kinase-5 (CDK5) and amyotrophic lateral sclerosis. Amyotroph Lateral Scler. Other Motor Neuron Disord 1:319–327
Beaulieu JM, Robertson J, Julien JP (1999) Interactions between peripherin and neurofilaments in cultured cells: disruption of peripherin assembly by the NF-M and NF-H subunits. Biochem Cell Biol 77:41–45
Bennett GS, Tapscott SJ, Kleinbart FA, Antin PB, Holtzer H (1981) Different proteins associated with 10-nanometer filaments in cultured chick neurons and nonneuronal cells. Science 212:567–569
Black MM, Lee VM (1988) Phosphorylation of neurofilament proteins in intact neurons: demonstration of phosphorylation in cell bodies and axons. J Neurosci 8:3296–3305
Böhmer F, Szedlacsek S, Tabernero L, Ostman A, den Hertog J (2013) Protein tyrosine phosphatase structure-function relationships in regulation and pathogenesis. FEBS J 280(2):413–431
Brettschneider J, Petzold A, Schottle D, Claus A, Riepe M, Tumani H (2006a) The neurofilament heavy chain (NfH) in the cerebrospinal fluid diagnosis of Alzheimer’s disease. Dement Geriatr Cogn Disord 21:291–295
Brettschneider J, Petzold A, Sussmuth SD, Ludolph AC, Tumani H (2006b) Axonal damage markers in cerebrospinal fluid are increased in ALS. Neurology 66:852–856
Butterfield DA, Abdul HM, Opii W, Newman SF, Joshi G, Ansari MA, Sultana R (2006) Pin1 in Alzheimer’s disease. J Neurochem 98(6):1697–1706
Collard JF, Cote F, Julien JP (1995) Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature 375:61–64
Cote F, Collard JF, Julien JP (1993) Progressive neuronopathy in transgenic mice expressing the human neurofilament heavy gene: a mouse model of amyotrophic lateral sclerosis. Cell 73:35–46
Driver JA, Lu KP (2010) Pin1: a new genetic link between Alzheimer’s disease, cancer and aging. Curr Aging Sci 3(3):158–165
Friede RL, Samorajski T (1970) Axon calibre related to neurofilaments and microtubules in sciatic nerve fibers of rats and mice. Anat Rec 167:379–388
Giasson BI, Mushynski WE (1997) Study of proline-directed protein kinases involved in phosphorylation of the heavy neurofilament subunit. J Neurosci 17:9466–9472
Goldstein ME, Sternberger NH, Sternberger LA (1987) Phosphorylation protects neurofilaments against proteolysis. J Neuroimmunol 14:149–160
Gou JP, Leterrier JF (1995) Possible involvement of ubiquitination in neurofilament degradation. Biochem Biophys Res Commun 217(2):529–538
Grant P, Pant HC (2000) Neurofilament protein synthesis and phosphorylation. J Neurocytol 29(11–12):843–872
Grant P, Zheng Y, Pant HC (2006) Squid (Loligo pealei) giant fiber system: a model for studying neurodegeneration and dementia? Biol Bull 210:318–333
Hamdane M, Dourlen P, Bretteville A, Sambo AV, Ferreira S, Ando K, Kerdraon O, Bégard S, Geay L, Lippens G, Sergeant N, Delacourte A, Maurage CA, Galas MC, Buée L (2006) Pin1 allows for differential tau dephosphorylation in neuronal cells. Mol Cell Neurosci 32(1–2):155–160
Holzer M, Gartner U, Stobe A, Hartig W, Gruschka H, Bruckner MK, Arendt T (2002) Inverse association of Pin1 and tau accumulation in Alzheimer’s disease hippocampus. Acta Neuropathol 104:471–481
Jaffe H, Sharma P, Grant P, Pant H (2001) Characterization of the phosphorylation sites of the squid (Loligo pealei) high-molecular-weight neurofilament protein from giant axon axoplasm. J Neurochem 76:1022–1031
Kesavapany S, Li BS, Amin N, Zheng YL, Grant P, Pant HC (2004) Neuronal cyclin-dependent kinase 5: role in nervous system function and its specific inhibition by the Cdk5 inhibitory peptide. Biochim Biophys Acta 1697(1–2):143–153
Kesavapany S, Patel V, Zheng YL, Pareek TK, Bjelogrlic M, Albers W, Amin N, Jaffe H, Gutkind JS, Strong MJ, Grant P, Pant HC (2007) Inhibition of Pin1 reduces glutamate-induced perikaryal accumulation of phosphorylated neurofilament-H in neurons. Mol Biol Cell 18(9):3645–3655
Landrieu I, Smet-Nocca C, Amniai L, Louis JV, Wieruszeski JM, Goris J, Janssens V, Lippens G (2011) Molecular implication of PP2A and Pin1 in the Alzheimer’s disease specific hyperphosphorylation of tau. PLoS ONE 6(6):e21521
Lee MK, Cleveland DW (1996) Neuronal intermediate filaments. Annu Rev Neurosci 19:187–217
Lee MK, Marszalek JR, Cleveland DW (1994) A mutant neurofilament subunit causes massive, selective motor neuron death: implications for the pathogenesis of human motor neuron disease. Neuron 13:975–988
Liou YC, Ryo A, Huang HK, Lu PJ, Bronson R, Fujimori F, Uchida T, Hunter T, Lu KP (2002) Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Proc Natl Acad Sci U S A 99:1335–1340
Liu F, Grundke-Iqbal I, Iqbal K, Gong CX (2005a) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 22:1942–1950
Liu F, Iqbal K, Grundke-Iqbal I, Rossie S, Gong CX (2005b) Dephosphorylation of tau by protein phosphatase 5: impairment in Alzheimer’s disease. J Biol Chem 280(3):1790–1796
Liu Q, Xie F, Alvarado-Diaz A, Smith MA, Moreira PI, Zhu X, Perry G (2011) Neurofilamentopathy in neurodegenerative diseases. Open Neurol J 5:58–62
Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP (1999) The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 399:784–788
Lu KP, Liou YC, Vincent I (2003) Proline-directed phosphorylation and isomerization in mitotic regulation and in Alzheimer’s disease. BioEssays 25(2):174–181
Nguyen MD, Lariviere RC, Julien JP (2001) Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron 30:135–147
Nixon RA (1993) The regulation of neurofilament protein dynamics by phosphorylation: clues to neurofibrillary pathobiology. Brain Pathol 3:29–38
Nixon RA, Shea TB (1992) Dynamics of neuronal intermediate filaments: a developmental perspective. Cell Motil Cytoskeleton 22:81–91
Nixon RA, Lewis SE, Mercken M, Sihag RK (1994) [32P]orthophosphate and [35S]methionine label separate pools of neurofilaments with markedly different axonal transport kinetics in mouse retinal ganglion cells in vivo. Neurochem Res 19:1445–1453
Pant HC (1988) Dephosphorylation of neurofilament proteins enhances their susceptibility to degradation by calpain. Biochem J 256:665–668
Pastorino L, Sun A, Lu PJ, Zhou XZ, Balastik M, Finn G, Wulf G, Lim J, Li SH, Li X, Xia W, Nicholson LK, Lu KP (2006) The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature 440(7083):528–534
Patrick GN, Zukerberg L, Nikolic M, de la Monte S (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402:615–622
Pei JJ, Grundke-Iqbal I, Iqbal K, Bogdanovic N, Winblad B, Cowburn RF (1997) Elevated protein levels of protein phosphatases PP-2A and PP-2B in astrocytes of Alzheimer’s disease temporal cortex. J Neural Transm 104(11–12):1329–1338
Pei JJ, Gong CX, Iqbal K, Grundke-Iqbal I, Wu QL, Winblad B, Cowburn RF (1998) Subcellular distribution of protein phosphatases and abnormally phosphorylated tau in the temporal cortex from Alzheimer’s disease and control brains. J Neural Transm 105(1):69–83
Petzold A (2005) Neurofilament phospho forms: surrogate markers for axonal injury, degeneration and loss. J Neurol Sci 233(1–2):183–198
Ramakrishnan P, Dickson DW, Davies P (2003) Pin1 colocalization with phosphorylated tau in Alzheimer’s disease and other tauopathies. Neurobiol Dis 14(2):251–264
Rudrabhatla P, Pant HC (2010) Phosphorylation-specific peptidyl-prolyl isomerization of neuronal cytoskeletal proteins by Pin1: implications for therapeutics in neurodegeneration. J Alzheimers Dis 19:389–403
Rudrabhatla P, Albers W, Pant HC (2009) Peptidyl-prolyl isomerase 1 regulates protein phosphatase 2A-mediated topographic phosphorylation of neurofilament proteins. J Neurosci 29(47):14869–14880
Rudrabhatla P, Grant P, Jaffe H, Strong MJ, Pant HC (2010) Quantitative phosphoproteomic analysis of neuronal intermediate filament proteins (NF-M/H) in Alzheimer’s disease by iTRAQ. FASEB J 24(11):4396–4407
Rudrabhatla P, Jaffe H, Pant HC (2011) Direct evidence of phosphorylated neuronal intermediate filament proteins in neurofibrillary tangles (NFTs): phosphoproteomics of Alzheimer’s NFTs. FASEB J 25(11):3896–3905
Ryo A, Togo T, Nakai T, Hirai A, Nishi M, Yamaguchi A, Suzuki K, Hirayasu Y, Kobayashi H, Perrem K, Liou YC, Aoki I (2006) The prolyl-isomerase Pin1 accumulates in the Lewy bodies of Parkinson’s disease and facilitates the formation of alpha-synuclein inclusions. J Biol Chem 281:4117–4125
Shea TB, Zheng YL, Ortiz D, Pant HC (2004a) Cyclin-dependent kinase 5 increases perikaryal neurofilament phosphorylation and inhibits neurofilament axonal transport in response to oxidative stress. J Neurosci Res 76:795–800
Shea TB, Yabe JT, Ortiz D, Pimenta A, Loomis P, Goldman RD, Amin N, Pant HC (2004b) Cdk5 regulates axonaltransport and phosphorylation of neurofilaments in cultured neurons. J Cell Sci 117(Pt 6):933–941
Shea TB, Chan WK, Kushkuley J, Lee S (2009) Organizational dynamics, functions, and pathobiological dysfunctions of neurofilaments. Results Probl Cell Differ 48:29–45
Shetty KT, Link WT, Pant HC (1993) cdc2-like kinase from rat spinal cord specifically phosphorylates KSPXK motifs in neurofilament proteins: isolation and characterization. Proc Natl Acad Sci USA 90:6844–6848
Sihag RK, Inagaki M, Yamaguchi T, Shea TB, Pant HC (2007) Role of phosphorylation on the structural dynamics and function of types III and IV intermediate filaments. Exp Cell Res 313:2098–2109
Sternberger NH, Sternberger LA, Ulrich J (1985) Aberrant neurofilament phosphorylation in Alzheimer disease. Proc Natl Acad Sci USA 82:4274–4276
Strack S, Westphal RS, Colbran RJ, Ebner FF, Wadzinski BE (1997) Protein serine/threonine phosphatase 1 and 2A associate with and dephosphorylate neurofilaments. Brain Res Mol Brain Res 49(1–2):15–28
Sundaram JR, Poore CP, Sulaimee NH, Pareek T, Asad AB, Rajkumar R, Cheong WF, Wenk MR, Dawe GS, Chuang KH, Pant HC, Kesavapany S (2013). Specific inhibition of p25/Cdk5 activity by the Cdk5 inhibitory peptide reduces neurodegeneration in vivo. J Neurosci 33(1):334–343
Tortarolo M, Veglianese P, Calvaresi N, Botturi A, Rossi C, Giorgini A, Migheli A, Bendotti C (2003) Persistent activation of p38 mitogen-activated protein kinase in a mouse model of familial amyotrophic lateral sclerosis correlates with disease progression. Mol Cell Neurosci 23:180–192
Toyoshima I, Kato K, Sugawara M, Wada C, Masamune O (1998) Kinesin accumulation in chick spinal axonal swellings with beta, beta’-iminodipropionitrile (IDPN) intoxication. Neurosci Lett 249:103–106
Veeranna S, Shetty KT, Link WT, Jaffe H, Wang J, Pant HC (1995) Neuronal cyclin-dependent kinase-5 phosphorylation sites in neurofilament protein (NF-H) are dephosphorylated by protein phosphatase 2A. J Neurochem 64:2681–2690
Veeranna S, Amin ND, Ahn NG, Jaffe H, Winters CA, Grant P, Pant HC (1998) Mitogen-activated protein kinases (Erk1, 2) phosphorylate Lys-Ser-Pro (KSP) repeats in neurofilament proteins NF-H and NF-M. J Neurosci 18(11):4008–40021
Veeranna S, Shetty KT, Takahashi M, Grant P, Pant HC (2000) Cdk5 and MAPK are associated with complexes of cytoskeletal proteins in rat brain. Brain Res Mol Brain Res 76:229–236
Veeranna S, Yang DS, Lee JH, Vinod KY, Stavrides P, Amin ND, Pant HC, Nixon RA (2011) Declining phosphatases underlie aging-related hyperphosphorylation of neurofilaments. Neurobiol Aging 32(11):2016–2029
Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VM (2001) PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Exp Neurol 168(2):402–412
Xiao S, McLean J, Robertson J (2006) Neuronal intermediate filaments and ALS: a new look at an old question. Biochim Biophys Acta 1762:1001–1012
Xu Z, Cork LC, Griffin JW, Cleveland DW (1993) Increased expression of neurofilament subunit NF-L produces morphological alternations that resemble the pathology of human motor neuron disease. Cell 73:23–33
Xu Z, Dong DL, Cleveland DW (1994) Neuronal intermediate filaments: new progress on an old subject. Curr Opin Neurobiol 4:655–661
Yuan A, Rao MV, Sasaki T, Chen Y, Kumar A, Veeranna LRK, Eyer J, Peterson AC, Julien JP, Nixon RA (2006) Alphainternexin is structurally and functionally associated with the neurofilament triplet proteins in the mature CNS. J Neurosci 26:10006–10019
Zheng YL, Li BS, Veeranna S, Pant HC (2003) Phosphorylation of the head domain of neurofilament protein (NF-M): a factor regulating topographic phosphorylation of NF-M tail domain KSP sites in neurons. J Biol Chem 278:24026–24032
Zheng YL, Kesavapany S, Gravell M, Hamilton RS, Schubert M, Amin N, Albers W, Grant P, Pant HC (2005) A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J 24(1):209–220
Zhou J, Wang H, Feng Y, Chen J (2010) Increased expression of cdk5/p25 in N2a cells leads to hyperphosphorylation and impaired axonal transport of neurofilament proteins. Life Sci 86:532–537
Zhu Q, Couillard-Despre′s S, Julien J-P (1997) Delayed maturation of regenerating myelinated axons in mice lacking neurofilaments. Exp Neurol 148:299–316
Acknowledgments
This research was supported by the Intramural Research Programs of the NIH, National Institute of Neurological Disorders and Stroke.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Binukumar, B.K., Shukla, V., Amin, N.D. et al. Topographic regulation of neuronal intermediate filaments by phosphorylation, role of peptidyl-prolyl isomerase 1: significance in neurodegeneration. Histochem Cell Biol 140, 23–32 (2013). https://doi.org/10.1007/s00418-013-1108-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00418-013-1108-7