




Abstract
Focal demyelinated plaques in white matter, which are the hallmark of multiple sclerosis pathology, only partially explain the patient's clinical deficits. We thus analysed global brain pathology in multiple sclerosis, focusing on the normal-appearing white matter (NAWM) and the cortex. Autopsy tissue from 52 multiple sclerosis patients (acute, relapsing-remitting, primary and secondary progressive multiple sclerosis) and from 30 controls was analysed using quantitative morphological techniques. New and active focal inflammatory demyelinating lesions in the white matter were mainly present in patients with acute and relapsing multiple sclerosis, while diffuse injury of the NAWM and cortical demyelination were characteristic hallmarks of primary and secondary progressive multiple sclerosis. Cortical demyelination and injury of the NAWM, reflected by diffuse axonal injury with profound microglia activation, occurred on the background of a global inflammatory response in the whole brain and meninges. There was only a marginal correlation between focal lesion load in the white matter and diffuse white matter injury, or cortical pathology, respectively. Our data suggest that multiple sclerosis starts as a focal inflammatory disease of the CNS, which gives rise to circumscribed demyelinated plaques in the white matter. With chronicity, diffuse inflammation accumulates throughout the whole brain, and is associated with slowly progressive axonal injury in the NAWM and cortical demyelination.
Introduction
Multiple sclerosis is the most common neurological disease in young adults in the Western world (Noseworthy et al., 2000). In most patients, the disease begins with episodes of neurological dysfunction followed by complete or partial remission—the relapsing/remitting form of the disease (RRMS)—and is later transformed into uninterrupted progression—the secondary progressive phase of the disease (SPMS; Weinshenker et al., 1989). Some patients begin with a progressive disease course [primary progressive multiple sclerosis (PPMS); Lublin and Reingold 1996, Confavreux et al., 2000]. In rare cases, fulminant disease may lead to death within months after the disease onset [Marburg's type of acute multiple sclerosis (AMS); Marburg 1906]. Relapse frequency in the early phase of the disease influences the time of onset of progression, however once a threshold of disability is reached, progression of disability is not affected by relapses either before or after the onset of the progressive phase (Confavreux et al., 2003; Pittock et al., 2004). Furthermore, during the progressive phase, the rate of clinical deterioration is similar between SPMS and PPMS patients (Confavreux et al., 2000, 2003). While immunomodulatory and immunosuppressive treatments are beneficial in the acute and relapsing stages, as these diminish the formation of new enhancing focal white matter lesions (WMLs) (Goodin et al., 2002), they have little effect in patients with progressive disease (Noseworthy et al., 2000; Leary et al., 2003). These data indicate that the pathogenesis of brain damage may be different between relapsing and progressive phases of multiple sclerosis.
Pathologically, multiple sclerosis is a chronic inflammatory disease of the central nervous system, which leads to focal plaques of demyelination in the central nervous system (Noseworthy et al., 2000). Longitudinal as well as cross sectional magnetic resonance studies show that the formation of new WMLs is often associated with leakage of the paramagnetic marker gadolinium–DTPA, and mainly occurs in the acute and relapsing stages of the disease (Cotton et al., 2003). In patients with progressive disease, and particularly in PPMS patients, new white matter and gadolinium-enhancing lesions are rare (Thompson et al., 1991, 1997), but there are progressive signal abnormalities in the normal-appearing white matter (NAWM) as well as a progressive loss of brain volume (Rovaris et al., 2001; Ingle et al., 2003). These diffuse changes have in part been explained by axonal destruction in the plaques followed by secondary Wallerian degeneration (Evangelou et al., 2000; Lovas et al., 2000; Bjartmar and Trapp, 2001). However, they may also develop independently from focal WMLs (Pelletier et al., 2003), since diffuse white matter damage and axonal loss can be very severe in spite of only few and small focal WMLs (Rovaris et al., 2001; Pelletier et al., 2003; Rocca et al., 2003).
By concentrating on focal WMLs, previous neuropathological studies did not find major differences between patients with acute, relapsing and progressive disease (Brück et al., 2002). Here we describe that in patients with SPMS or PPMS, the whole brain is affected, as reflected by global inflammation, extensive cortical demyelination and diffuse axonal injury in the NAWM.
Materials and methods
Cases and autopsy material
This study was performed on archival autopsies from 52 multiple sclerosis cases, 15 cases of advanced Alzheimer's disease (Braak stages V and VI, Braak and Braak, 1991) and 15 normal controls, which were formalin fixed and paraffin embedded. Clinical histories were available on all of the cases, and clinical course was determined by retrospective chart review performed either by a neurologist or a neuropathologist (H.R., C.S., M.B. and C.F.L.) blinded to the pathological analysis. Clinical course was defined according to established criteria (Lublin and Reingold, 1996). Patients with AMS died within a year after the onset of the disease (Marburg, 1906). Eleven AMS cases, 6 RRMS cases, 15 PPMS cases and 20 SPMS cases were included in the study (Table 1). Diagnosis of multiple sclerosis was histologically confirmed by a neuropathologist (M.S., W.B., H.L., J.E.P.).
Quantitative differences in pathological features between different subgroups of multiple sclerosis values
| AMS | RRMS | SPMS | PPMS | Controls | Alzheimer's disease | P-value group comparison | P-value pooled course | |
|---|---|---|---|---|---|---|---|---|
| Number of cases | 11 | 6 | 20 | 14 | 15 | 15 | ||
| Mean age (years); range | 45.27 (28–68) | 50.5 (20–67) | 46.4 (28–61) | 53.9 (28–75) | 74.6 (46–89) | 79.7 (60–92) | ||
| Female/male ratio | 1.75 | 1 | 1.44 | 1.33 | 1.14 | 1 | ||
| Disease duration (months) | 1.5 (0.2–7) | 120 (48–156) | 192 (72–408) | 198 (30–411) | 0 | n.a. | P < 0.001 | P < 0.001 |
| WML area forebrain (%) | 22.66 (0–85.05) | 10.3(1.01–53.25) | 24.13 (2.79–60.36) | 6.54 (0.46 –76.54) | 0 | Atrophy | P = 0.06 | P = 0.78 |
| Cortical lesion area forebrain (%) | 0 (0–3.93) | 2.96 (0–14.14) | 13.29 (0–68.63) | 12.54 (0–38.68) | 0 | Plaques + tangles | P < 0.001 | P < 0.001 |
| Active WMLs (%) | 100 (90–100) | 11.44 (0–100) | 0 (0–50) | 0 (0–56.25) | 0 | n.a. | P < 0.001 | P < 0.001 |
| Slowly expanding WMLs (%) | 0 (0) | 10.42 (0–100) | 14.29 (0–53.85) | 12.5 (0–50) | 0 | n.a. | P < 0.001 | P < 0.004 |
| Inactive WMLs (%) | 0 (0–10) | 35.3 (0–85.71) | 85.71 (12.5–100) | 77.78 (14.29–100) | 0 | n.a. | P < 0.001 | P < 0.001 |
| Inflammatory infiltrates meninges (per 100 mm) | 0.56 (0.196–2.793) | 0.42 (0–0.824) | 0.86 (0–4.752) | 0.64 (0–4.506) | 0 | 0 | P = 0.58 | P = 0.3 |
| Perivascular inflammatory infiltrates NAWM (per mm2) | 0.04 (0.015–0.12) | 0.05 (0.03–0.21) | 0.27 (0.015–0.79) | 0.13 (0.015–0.88) | 0 | 0.009 (0.0–0.03) | P < 0.001 | P < 0.004 |
| Microglia activation NAWM | 1.5 (1–4) | 2 (2–4) | 4 (2–5) | 4 (2–5) | 1 (1) | 2 (1–4) | P < 0.001 | P < 0.001 |
| Axonal spheroids NAWM (per mm2) | 2.69 (0–7) | 4.67 (1.65–7.5) | 10.25 (0–35.06) | 16.8 (2.5–81.5) | 0.5 (0–5.75) | 3.775 (0.78–7.8) | P < 0.001 | P < 0.001 |
| AMS | RRMS | SPMS | PPMS | Controls | Alzheimer's disease | P-value group comparison | P-value pooled course | |
|---|---|---|---|---|---|---|---|---|
| Number of cases | 11 | 6 | 20 | 14 | 15 | 15 | ||
| Mean age (years); range | 45.27 (28–68) | 50.5 (20–67) | 46.4 (28–61) | 53.9 (28–75) | 74.6 (46–89) | 79.7 (60–92) | ||
| Female/male ratio | 1.75 | 1 | 1.44 | 1.33 | 1.14 | 1 | ||
| Disease duration (months) | 1.5 (0.2–7) | 120 (48–156) | 192 (72–408) | 198 (30–411) | 0 | n.a. | P < 0.001 | P < 0.001 |
| WML area forebrain (%) | 22.66 (0–85.05) | 10.3(1.01–53.25) | 24.13 (2.79–60.36) | 6.54 (0.46 –76.54) | 0 | Atrophy | P = 0.06 | P = 0.78 |
| Cortical lesion area forebrain (%) | 0 (0–3.93) | 2.96 (0–14.14) | 13.29 (0–68.63) | 12.54 (0–38.68) | 0 | Plaques + tangles | P < 0.001 | P < 0.001 |
| Active WMLs (%) | 100 (90–100) | 11.44 (0–100) | 0 (0–50) | 0 (0–56.25) | 0 | n.a. | P < 0.001 | P < 0.001 |
| Slowly expanding WMLs (%) | 0 (0) | 10.42 (0–100) | 14.29 (0–53.85) | 12.5 (0–50) | 0 | n.a. | P < 0.001 | P < 0.004 |
| Inactive WMLs (%) | 0 (0–10) | 35.3 (0–85.71) | 85.71 (12.5–100) | 77.78 (14.29–100) | 0 | n.a. | P < 0.001 | P < 0.001 |
| Inflammatory infiltrates meninges (per 100 mm) | 0.56 (0.196–2.793) | 0.42 (0–0.824) | 0.86 (0–4.752) | 0.64 (0–4.506) | 0 | 0 | P = 0.58 | P = 0.3 |
| Perivascular inflammatory infiltrates NAWM (per mm2) | 0.04 (0.015–0.12) | 0.05 (0.03–0.21) | 0.27 (0.015–0.79) | 0.13 (0.015–0.88) | 0 | 0.009 (0.0–0.03) | P < 0.001 | P < 0.004 |
| Microglia activation NAWM | 1.5 (1–4) | 2 (2–4) | 4 (2–5) | 4 (2–5) | 1 (1) | 2 (1–4) | P < 0.001 | P < 0.001 |
| Axonal spheroids NAWM (per mm2) | 2.69 (0–7) | 4.67 (1.65–7.5) | 10.25 (0–35.06) | 16.8 (2.5–81.5) | 0.5 (0–5.75) | 3.775 (0.78–7.8) | P < 0.001 | P < 0.001 |
These values represent medians, the range for values are given in brackets; n.a.: not applicable.
Values in bold indicate those contributing to the significant differences.
Quantitative differences in pathological features between different subgroups of multiple sclerosis values
| AMS | RRMS | SPMS | PPMS | Controls | Alzheimer's disease | P-value group comparison | P-value pooled course | |
|---|---|---|---|---|---|---|---|---|
| Number of cases | 11 | 6 | 20 | 14 | 15 | 15 | ||
| Mean age (years); range | 45.27 (28–68) | 50.5 (20–67) | 46.4 (28–61) | 53.9 (28–75) | 74.6 (46–89) | 79.7 (60–92) | ||
| Female/male ratio | 1.75 | 1 | 1.44 | 1.33 | 1.14 | 1 | ||
| Disease duration (months) | 1.5 (0.2–7) | 120 (48–156) | 192 (72–408) | 198 (30–411) | 0 | n.a. | P < 0.001 | P < 0.001 |
| WML area forebrain (%) | 22.66 (0–85.05) | 10.3(1.01–53.25) | 24.13 (2.79–60.36) | 6.54 (0.46 –76.54) | 0 | Atrophy | P = 0.06 | P = 0.78 |
| Cortical lesion area forebrain (%) | 0 (0–3.93) | 2.96 (0–14.14) | 13.29 (0–68.63) | 12.54 (0–38.68) | 0 | Plaques + tangles | P < 0.001 | P < 0.001 |
| Active WMLs (%) | 100 (90–100) | 11.44 (0–100) | 0 (0–50) | 0 (0–56.25) | 0 | n.a. | P < 0.001 | P < 0.001 |
| Slowly expanding WMLs (%) | 0 (0) | 10.42 (0–100) | 14.29 (0–53.85) | 12.5 (0–50) | 0 | n.a. | P < 0.001 | P < 0.004 |
| Inactive WMLs (%) | 0 (0–10) | 35.3 (0–85.71) | 85.71 (12.5–100) | 77.78 (14.29–100) | 0 | n.a. | P < 0.001 | P < 0.001 |
| Inflammatory infiltrates meninges (per 100 mm) | 0.56 (0.196–2.793) | 0.42 (0–0.824) | 0.86 (0–4.752) | 0.64 (0–4.506) | 0 | 0 | P = 0.58 | P = 0.3 |
| Perivascular inflammatory infiltrates NAWM (per mm2) | 0.04 (0.015–0.12) | 0.05 (0.03–0.21) | 0.27 (0.015–0.79) | 0.13 (0.015–0.88) | 0 | 0.009 (0.0–0.03) | P < 0.001 | P < 0.004 |
| Microglia activation NAWM | 1.5 (1–4) | 2 (2–4) | 4 (2–5) | 4 (2–5) | 1 (1) | 2 (1–4) | P < 0.001 | P < 0.001 |
| Axonal spheroids NAWM (per mm2) | 2.69 (0–7) | 4.67 (1.65–7.5) | 10.25 (0–35.06) | 16.8 (2.5–81.5) | 0.5 (0–5.75) | 3.775 (0.78–7.8) | P < 0.001 | P < 0.001 |
| AMS | RRMS | SPMS | PPMS | Controls | Alzheimer's disease | P-value group comparison | P-value pooled course | |
|---|---|---|---|---|---|---|---|---|
| Number of cases | 11 | 6 | 20 | 14 | 15 | 15 | ||
| Mean age (years); range | 45.27 (28–68) | 50.5 (20–67) | 46.4 (28–61) | 53.9 (28–75) | 74.6 (46–89) | 79.7 (60–92) | ||
| Female/male ratio | 1.75 | 1 | 1.44 | 1.33 | 1.14 | 1 | ||
| Disease duration (months) | 1.5 (0.2–7) | 120 (48–156) | 192 (72–408) | 198 (30–411) | 0 | n.a. | P < 0.001 | P < 0.001 |
| WML area forebrain (%) | 22.66 (0–85.05) | 10.3(1.01–53.25) | 24.13 (2.79–60.36) | 6.54 (0.46 –76.54) | 0 | Atrophy | P = 0.06 | P = 0.78 |
| Cortical lesion area forebrain (%) | 0 (0–3.93) | 2.96 (0–14.14) | 13.29 (0–68.63) | 12.54 (0–38.68) | 0 | Plaques + tangles | P < 0.001 | P < 0.001 |
| Active WMLs (%) | 100 (90–100) | 11.44 (0–100) | 0 (0–50) | 0 (0–56.25) | 0 | n.a. | P < 0.001 | P < 0.001 |
| Slowly expanding WMLs (%) | 0 (0) | 10.42 (0–100) | 14.29 (0–53.85) | 12.5 (0–50) | 0 | n.a. | P < 0.001 | P < 0.004 |
| Inactive WMLs (%) | 0 (0–10) | 35.3 (0–85.71) | 85.71 (12.5–100) | 77.78 (14.29–100) | 0 | n.a. | P < 0.001 | P < 0.001 |
| Inflammatory infiltrates meninges (per 100 mm) | 0.56 (0.196–2.793) | 0.42 (0–0.824) | 0.86 (0–4.752) | 0.64 (0–4.506) | 0 | 0 | P = 0.58 | P = 0.3 |
| Perivascular inflammatory infiltrates NAWM (per mm2) | 0.04 (0.015–0.12) | 0.05 (0.03–0.21) | 0.27 (0.015–0.79) | 0.13 (0.015–0.88) | 0 | 0.009 (0.0–0.03) | P < 0.001 | P < 0.004 |
| Microglia activation NAWM | 1.5 (1–4) | 2 (2–4) | 4 (2–5) | 4 (2–5) | 1 (1) | 2 (1–4) | P < 0.001 | P < 0.001 |
| Axonal spheroids NAWM (per mm2) | 2.69 (0–7) | 4.67 (1.65–7.5) | 10.25 (0–35.06) | 16.8 (2.5–81.5) | 0.5 (0–5.75) | 3.775 (0.78–7.8) | P < 0.001 | P < 0.001 |
These values represent medians, the range for values are given in brackets; n.a.: not applicable.
Values in bold indicate those contributing to the significant differences.
Neuropathology and immunohistochemistry
Neuropathological analysis was performed either on large hemispheric or double hemispheric brain sections (117 tissue blocks, from 31 multiple sclerosis cases and 3 Alzheimer's disease cases), or on multiple sections from different brain areas (a total of 398 tissue blocks). For basic classification of inflammation, demyelination and diffuse white matter injury, sections were stained with haematoxylin and eosin (H&E), Luxol fast blue myelin stain and Bielschowsky silver impregnation. Immunocytochemistry was performed using a biotin avidin technique (Bien et al., 2002), with antibodies against the following targets: CD3 (T-cells; Serotec, Oxford, UK); CD8 (Class I MHC restricted T-cells; Dako, Glostrup, DK), CD68 (macrophages and microglia; Dako, Glostrup, DK), proteolipid protein (PLP, myelin, Serotec, Oxford, UK) and neurofilament (axons, Chemicon, Temecula, CA, USA).
Quantitative analysis
All quantitative data were obtained by one investigator blinded to the clinical data (A.K.). All sections were scanned and a camera lucida image of each entire section was drawn. In these scans, demyelinated areas in the white matter (identified by Luxol fast blue myelin stain) and grey matter [seen in sections stained by immunocytochemistry for PLP (Peterson et al., 2001); Fig. 1M and N] were manually outlined (Fig. 1). The templates were then overlaid by a morphometric grid and the area of demyelinated and normal tissue was determined. The values in Table 1 represent the median percentage of demyelinated cortex and white matter in relation to the total cortical and white matter area, respectively.
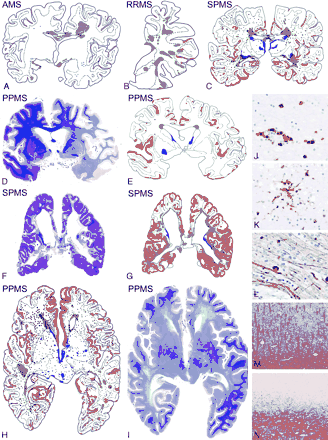
Focal inflammatory demyelinated plaques in the white matter dominate the pathology in acute and relapsing multiple sclerosis, while cortical demyelination and diffuse white matter inflammation are characteristic hallmarks of PPMS and SPMS. A, B, C, E, G and H: schematic lesion maps of multiple sclerosis brains; green: focal demyelinated plaques in the white matter; red: cortical demyelination; blue: demyelinated lesions in the deep grey matter; dark blue dots: inflammatory infiltrates in the brain; light blue dots in C, E, G and H: inflammatory infiltrates in the meninges. A: AMS: male, aged 35 years, 1.5 months disease duration; B: RRMS: female, aged 57 years, 13 years disease duration; C: SPMS: male, 43 years, 16 years disease duration; D and E: PPMS with severe demyelination in the cortex but only minor disease involvement of the white matter, Luxol fast blue stain and corresponding scheme, ×0.3; F and G: SPMS: female, aged 46 years, 16 years disease duration, showing extensive damage in both cortex and white matter, Luxol fast blue stain and corresponding scheme, × 0.3; H: PPMS: female, aged 55 years, 5 years disease duration; I: diffuse white matter abnormalities in PPMS; only the subcortical myelin is intact; there are only few focal demyelinated plaques (see also H); Luxol fast blue stain, ×0.5; J: inflammation in the NAWM in PPMS; immunocytochemistry for CD8, ×150; K: microglia activation and formation of microglia nodules in the NAWM of PPMS, ×75; L: diffuse axonal injury, reflected by axonal swellings and end bulbs in the NAWM of PPMS, immunocytochemistry for neurofilament, ×150; M: normal cortical myelin in a patient with SPMS; immunocytochemistry for PLP, × 12; N: adjacent cortical area to that shown in M with complete demyelination in the entire cortex; myelin in the subcortical white matter is intact; immunocytochemistry for PLP, ×12.
In addition, a detailed quantitative evaluation of inflammation, microglia activation and axonal injury was performed in randomly sampled areas of the NAWM, which were at least at 1 cm distance from focal demyelinated plaques and outside fibre tracts emerging from WMLs. On an average 4 blocks per case (3 × 2 cm) were analysed when only small blocks were available. The blocks were randomly sampled at autopsy and contained tissue with multiple sclerosis plaques as well as others from macroscopically normal white matter. Perivascular inflammatory infiltrates were counted on H&E stained sections in multiple randomly sampled low power fields (total area of 180 mm2/case) and the values expressed as the mean number of inflammatory infiltrates per mm2. For meningeal inflammation, the exact location of perivascular infiltrates in the meninges was drawn into the camera lucida templates. The density of infiltrates was determined and expressed as that present in 100 mm of meninges per case. The presence of macrophages and microglia activation was evaluated on an average in three sections per case in the NAWM. Sections stained by immunocytochemistry for CD68 were analysed using a semi-quantitative scoring system (0: no CD68 expression; 1: scattered CD68 positive microglia cells; 2: most microglia cells in the tissue with CD68 reactivity). In addition the presence or absence of microglia nodules (Fig. 1K) was also determined (0: no microglia nodules; 1: presence of microglia nodules). Tissue macrophages were defined as round cells with foamy cytoplasm and strong CD68 reactivity. Semi-quantitative scoring was defined by 0: no macrophages, 1: few perivascular macrophages and 2: macrophages dispersed within the tissue. The values in the figures and tables represent average summary scores per patient (microglia score + presence of microglia nodules + macrophage score; a total maximal value of 5).
Axonal injury was defined by the presence of focal axonal swellings and axonal end bulbs, visualized in sections stained by immunocytochemistry for neurofilament (Trapp et al., 1998) (Fig. 1L). Their density was determined using an ocular morphometric grid in 80 high power fields per each individual section, irrespective of being double hemispheric, hemispheric or smaller sections, representing a total area of 5 mm2 per case. Values are expressed as the number of injured axons per mm2.
To evaluate plaque activity, a total of 980 focal demyelinated white matter plaques were analysed. Each individual plaque was classified by two independent investigators (A.K. and H.L.; inter-rater agreement: 100%) into the following categories: classical actively demyelinating plaques were defined by the presence of abundant macrophages with early myelin degradation products either throughout the whole lesion or at the lesion edge (Brück et al., 1995); slowly expanding chronic plaques revealed mild to moderate microglia activation and macrophage infiltration at the lesion margins, early myelin degradation products, however, were sparse and restricted to single macrophages (Prineas et al., 2001); and inactive plaques showed a sharp lesion edge devoid of macrophage infiltration.
Statistical analysis
In a first step, global group differences between different courses of multiple sclerosis and controls were analysed for all quantitative parameters described above. In a second step, the values for AMS and RRMS as well as those for PPMS and SPMS, respectively, were pooled and the two groups were compared. For the analysis, non-parametric group tests (Kruskal–Wallis) were used. Spearman's rank correlations were used to identify interdependence of variables. P-values < 0.004 were regarded as significant after correction for multiple testing (Bonferroni).
Results
Active white matter plaques are mainly present in acute and relapsing multiple sclerosis
Focal WML load in the forebrain was not significantly different between the four groups, although it was slightly higher in AMS and SPMS and lowest in PPMS (Table 1). However, major differences were found in the activity patterns of focal WMLs (P < 0.001; Table 1). Patients with AMS revealed the highest incidence of classical active lesions, followed by patients with RRMS. The number of classical active lesions was low in patients with progressive disease, but on an average 13% of the plaques showed slow radial expansion in patients with RRMS, SPMS and PPMS.
Cortical demyelination is a characteristic feature of progressive multiple sclerosis
The amount of cortical demyelination was strikingly different in relation to the disease course. Demyelination in the cerebral cortex was mainly a feature in patients with SPMS and PPMS, but was rare or absent in patients with acute or relapsing disease (Fig. 1; Table 1). Demyelination mainly affected the subpial layers of the cerebral cortex and was associated with mononuclear inflammatory infiltrates in the meninges.
Inflammation is focal in acute and relapsing multiple sclerosis but global in progressive multiple sclerosis
Consistent with previous descriptions of multiple sclerosis pathology, we found the most extensive inflammatory reaction in focal white matter plaques (data not shown). Inflammation was most severe in classical active lesions, followed in the order of magnitude by lesions with slow expansion at the lesion edges and inactive plaques. However, there was a mild, but diffuse inflammatory reaction in the NAWM of patients with SPMS and PPMS, which was significantly less pronounced in patients with acute and relapsing multiple sclerosis (P = 0.003; Fig. 1; Table 1). This diffuse inflammatory reaction consisted of perivascular cuffs of mononuclear cells and a diffuse infiltration of the tissue by T-lymphocytes (Fig. 1J). This inflammatory process was associated with profound microglia activation. Microglia activation was reflected by an increased density of CD68 positive cells and the formation of microglia nodules (Fig. 1K), which were significantly more frequent in the NAWM in patients with progressive disease than in patients with acute or relapsing disease (P < 0.001). Meningeal inflammation was present in all the disease stages.
Progressive multiple sclerosis is associated with diffuse injury in the NAWM
Widespread and diffuse injury was present in the NAWM in patients with SPMS and PPMS (Fig. 1). This was most clearly evident when whole hemispheric or double hemispheric sections were analysed, and consisted of a global reduction in the intensity of myelin staining due to decreased fibre density (axons and myelin), which generally spared the subcortical U-fibres (Fig. 1I). Primary demyelination was largely absent. In contrast, focal axonal swellings and axonal end bulbs were frequently found in sections stained by immunocytochemistry for neurofilaments (Fig. 1L). Degenerating axons were present throughout the whole white matter, although there was some increase around demyelinated plaques and within defined fibre tracts emerging from the plaques. Patients with progressive disease showed significantly more diffuse axonal injury in the NAWM compared with patients with acute or relapsing disease (P < 0.001; Table 1).
Diffuse white matter injury in part correlates with cortical demyelination but not with focal WMLs
We found a significant correlation between the extent of cortical demyelination in the forebrain, with global inflammation and microglia activation in the NAWM (P = 0.01; r = 0.41 for inflammation and P = 0.001; r = 0.55 for microglia activation). Only a weak correlation between the extent of demyelination in the white matter and diffuse inflammation in the NAWM was found (P = 0.01; r = 0.29). No significant correlation was present between the focal WML load and microglia activation or axonal injury in the NAWM, or between plaque load in the white matter and cortical demyelination.
The view that cortical plaques and diffuse white matter injury develop independently from WMLs was further supported by the analysis of individual cases. Some patients showed extensive plaque like demyelination in the white matter with very little involvement of the NAWM and the cortex (Fig. 1B), while in others cortical demyelination was severe, in spite of very few WMLs (Fig. 1D and E). Other patients revealed massive cortical demyelination and extensive injury in the NAWM with only few white matter plaques (Fig. 1H and I). Finally in some patients all lesion types coincided leading to extensive focal and diffuse brain damage (Fig. 1F and G).
Influences of age, gender and disease duration on pathological changes
As expected, we found a highly significant correlation between disease duration and plaque activity (P = 0.001, r = 0.74), with classical active plaques being mainly present in patients with short disease duration. No correlation was found between any of the other pathologies described above with disease duration, age or gender of the patients. No significant differences in brain lesion load or inflammation, microglia activation and axonal injury were found between patients, dying from infectious (septic) complications or vascular disease.
Differences between SPMS and PPMS
Patients with PPMS showed a lower, but not significantly different load of focal demyelinated plaques in the white matter in comparison with those with SPMS (Table 1). As described before for focal demyelinated plaques (Revesz et al., 1994), we found lower numbers of inflammatory infiltrates also in the global white matter in PPMS compared with SPMS patients. There was no difference in the extent of diffuse white matter injury, diffuse microglia activation and cortical demyelination.
Inflammation and neurodegeneration in Alzheimer cases and normal controls
There was only a modest level of inflammation and neurodegeneration in the control groups (Table 1). While only very few axonal spheroids were seen in patients dying from infectious or vascular diseases, axonal damage in the white matter of the Alzheimer cases was more frequent, but not significantly lower compared with that in patients with multiple sclerosis. Only exceptional perivascular inflammatory infiltrates were present in the white matter of Alzheimer's disease patients. The activation of microglia cells in the white matter of the normal controls was mild and although microglia activation was more pronounced in the white matter of the Alzheimer cases, there was no formation of microglia nodules and no macrophages were seen within the tissue. No correlation was found between inflammation, or microglia activation, and axonal damage in Alzheimer's disease patients.
Discussion
This study defines for the first time the pathological differences between patients with progressive multiple sclerosis in comparison with patients with acute or relapsing disease. According to our results, brains of people with multiple sclerosis are affected by three different pathologies, which dominate during different stages of the disease. Focal demyelinated plaques are present in all stages of the disease, however classical active plaques are predominantly formed in patients with acute or relapsing disease, while focal white matter plaques in progressive multiple sclerosis were either inactive or showed slow expansion on their edges. In contrast, cortical demyelination and diffuse white matter injury are most prominent in patients with PPMS or SPMS, being rare or absent in the acute or relapsing stage.
Our data suggest that these three different pathological processes may at least in part develop independently from each other. We found that diffuse white matter injury can be profound even in patients with very low total brain lesion load. Similar conclusions have recently been reported in MRI studies (Pelletier et al., 2003; Rocca et al., 2003). Although we observed a significant correlation between the extent of demyelination in the cortex and diffuse white matter injury, the correlation coefficient was low. Furthermore, since cortical demyelination and diffuse white matter injury occurred in the same stage of the disease, this correlation could reflect a stage dependent common pathogenetic pathway of tissue injury rather than a true interdependence. This partly independent development of these three different pathologies may give rise to distinct disease patterns, in which either cortical demyelination, focal white matter plaques or diffuse injury in the NAWM dominates.
Previous studies have been limited in their ability to correlate functional neurological deficit with focal WMLs in the brain or spinal cord, determined by quantitative MRI techniques (Kidd et al., 1993, 1996; Rovaris et al., 2001; Ingle et al., 2003). Furthermore, recent MRI data suggest that permanent neurological deficit may in part be attributed to global and diffuse changes in the NAWM (Ciccarelli et al., 2001; Rovaris et al., 2001, 2002; Dehmeshki et al., 2003). Our recent data show profound axonal injury in the white matter in patients with progressive multiple sclerosis. This is well reflected in studies using magnetic resonance spectroscopy, which revealed a progressive reduction of N-acetyl aspartate in the global white matter in both PPMS and SPMS (Fu et al., 1998; De Stefano et al., 1999; Leary et al., 1999; Sarchielli et al., 1999).
Our study, however, highlights cortical pathology as another possible culprit for neurological dysfunction in progressive multiple sclerosis patients. The presence of cortical demyelination in patients with SPMS and PPMS has been previously recognized (Brownell and Hughes 1962; Peterson et al., 2001), but it is rare in the early stages of multiple sclerosis. Furthermore, cortical damage can be extensive, as seen in the analysis of double hemispheric sections, affecting up to 68% of the total forebrain cortical area in some extreme examples. The functional consequences of demyelination in the cerebral cortex are not yet defined, but MRI data suggest that cortical damage and atrophy may indeed be clinically significant (Richert et al., 1998; Dehmeshki et al., 2003; De Stefano et al., 2003).
It is important to note that all diffuse white matter injury and cortical demyelination in progressive multiple sclerosis invariably occurred on a background of meningeal, perivascular and parenchymal inflammation. It has been noted before that profound microglia activation with the formation of microglia nodules is present in the periplaque white matter of patients with progressive multiple sclerosis, but not around acute plaques (Prineas et al., 2001). Our study shows that this is not restricted to the area around plaques but affects the NAWM in a global sense. The inflammatory nature of diffuse white matter injury in patients with progressive multiple sclerosis has recently been challenged in MRI studies, which show that this type of damage occurs in the absence of contrast enhancement (Thompson et al., 1991; Silver et al., 2001). Furthermore, the lack of efficacy of immunomodulatory and immunosuppressive therapies in patients with progressive disease (Noseworthy et al., 2000; Leary et al., 2003) is regarded as indicative of the fact that neurodegeneration in such patients occurs independent of inflammation. However, contrast enhancement in MRI is not a marker for presence or absence of inflammation, but it rather detects evidence for blood–brain barrier damage, which is associated with transmigration of inflammatory cells through the wall of cerebral vessels. Furthermore its sensitivity for determining blood–brain barrier damage is low. For these reasons it seems to be a reliable technique to visualize massive new waves of inflammation, which are associated with the formation of new focal plaques in the CNS of multiple sclerosis patients, but may be insufficient to detect more subtle blood–brain barrier alterations, which may occur in the course of a gradual accumulation of inflammatory cells (Silver et al., 2001). Recently it was shown that in SPMS the inflammatory reaction leads to the formation of lymphatic like structures within the meninges and the perivascular spaces (Serafini et al., 2004). This observation is consistent with the view that in this stage of the disease the inflammatory reaction becomes trapped behind an intact or repaired blood–brain barrier. Such a compartmentalized inflammatory reaction is also unlikely to respond to current immunosuppressive or immunomodulatory treatments.
We have previously reported that the pathology of classical active lesions in early stages of the disease is heterogeneous (Lucchinetti et al., 2000). To what extent a similar heterogeneity is also present in active lesions in patients with late progressive disease is so far unknown. Whether or not the immunopathological patterns we have previously described in classical active multiple sclerosis lesions during acute or relapsing multiple sclerosis impact the diffuse white matter or cortical pathology observed during the progressive course of the disease has to be determined.
In conclusion, our data are consistent with the view that distinct pathogenetic processes, which in part may develop independently from each other, mediate brain damage in multiple sclerosis patients. New waves of inflammation, which enter the CNS from the circulation, appear to be responsible for the formation of focal plaques of demyelination, which are preferentially located within the white matter and seem to be mainly, but not exclusively, responsible for tissue damage in patients with acute and relapsing multiple sclerosis. However, with chronicity of the disease a slow but continuous accumulation of inflammatory cells within the whole brain may occur, which escapes the control of the peripheral immune system. This diffuse inflammatory process may then lead in a slowly progressive process to global damage within the white matter and, due to its chronic presence in the meninges, to subpial demyelination and tissue damage in the cerebral cortex. Due to the limited number of patients with RRMS in our present study we could not determine the time of progression of this process in the natural course of multiple sclerosis. However, MRI studies reveal the evidence for diffuse white matter damage being present in some patients, very early in the disease course (De Stefano et al., 2002; Filippi et al., 2003).
The authors thank Marianne Leiszer and Ulrike Köck for their expert technical assistance. This study was supported by the Fonds zur Förderung der wissenschaftlichen Forschung, Austria (grant P 16848-B02), by a research prize from the Roman, Marga und Marielle Sobek Stiftung, and by the US National Multiple Sclerosis Society (grant RG 3051-A-1). W.B. and C.S. are supported by the Gemeinnützige Hertie-Stiftung. C.S. is furthermore supported by the medical faculty of the University of Göttingen (junior research group).
References
Bien CG, Bauer J, Deckwerth TL, Wiendl H, Deckert M, Wiestler OD, et al. Destruction of neurons by cytotoxic T cells: a new pathogenic mechanism in Rasmussen's encephalitis.
Bjartmar C, Trapp BD. Axonal and neuronal degeneration in multiple sclerosis: mechanisms and functional consequences.
Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes.
Brownell B, Hughes JT. The distribution of plaques in the cerebrum in multiple sclerosis.
Brück W, Porada P, Poser S, Rieckmann P, Hanefeld F, Kretzschmar HA, et al. Monocyte/macrophage differentiation in early multiple sclerosis.
Brück W, Lucchinetti C, Lassmann H. The pathology of primary progressive multiple sclerosis.
Ciccarelli O, Werring DJ, Wheeler-Kingshott CA, Barker GJ, Parker GJ, Thompson AJ, et al. Investigation of multiple sclerosis normal-appearing brain using diffusion tensor MRI with clinical correlations.
Confavreux C, Vukusic S, Moreau T, Adeleine P. Relapses and progression of disability in multiple sclerosis.
Confavreux C, Vukusic S, Adeleine P. Early clinical predictors and progression of irreversible disability in multiple sclerosis: an amnesic process.
Cotton F, Weiner HL, Jolesz FA, Guttmann CR. MRI contrast uptake in new lesions in relapsing-remitting multiple sclerosis followed at weekly intervals.
De Stefano N, Narayanan S, Matthews PM, Francis GS, Antel JP, Arnold DL. In vivo evidence for axonal dysfunction remote from focal cerebral demyelination of the type seen in multiple sclerosis.
De Stefano N, Narayanan S, Francis SJ, Smith S, Mortilla M, Tartaglia MC, et al. Diffuse axonal and tissue injury in patients with multiple sclerosis with low cerebral lesion load and no disability.
De Stefano N, Matthews PM, Filippi M, Agosta F, De Luca M, Bartolozzi ML, et al. Evidence of early cortical atrophy in multiple sclerosis: relevance to white matter changes and disability.
Dehmeshki J, Chard DT, Leary SM, Watt HC, Silver NC, Tofts PS, et al. The normal appearing grey matter in primary progressive multiple sclerosis: a magnetisation transfer imaging study.
Evangelou N, Konz D, Esiri MM, Smith S, Palace J, Matthews PM. Regional axonal loss in the corpus callosum correlates with cerebral white matter lesion volume and distribution in multiple sclerosis.
Filippi M, Bozzali M, Rovaris M, Gonen O, Kesavadas C, Ghezzi A, et al. Evidence for widespread axonal damage at the earliest clinical stage of multiple sclerosis.
Fu L, Matthews PM, De Stefano N, Worsley KJ, Narayanan S, Francis GS, et al. Imaging axonal damage of normal-appearing white matter in multiple sclerosis.
Goodin DS, Frohman EM, Garmany GP Jr, Halper J, Likosky WH, Lublin FD, et al. Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines. Disease modifying therapies in multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology and the MS Council for Clinical Practice Guidelines.
Ingle GT, Stevenson VL, Miller DH, Thompson AJ. Primary progressive multiple sclerosis: a 5-year clinical and MR study.
Kidd D, Thorpe JW, Thompson AJ, Kendall BE, Moseley IF, MacManus DG, et al. Spinal cord MRI using multi-array coils and fast spin echo. II. Findings in multiple sclerosis.
Kidd D, Thorpe JW, Kendall BE, Barker GJ, Miller DH, McDonald WI, et al. MRI dynamics of brain and spinal cord in progressive multiple sclerosis.
Leary SM, Davie CA, Parker GJ, Stevenson VL, Wang L, Barker GJ, et al. 1H magnetic resonance spectroscopy of normal appearing white matter in primary progressive multiple sclerosis.
Leary SM, Miller DH, Stevenson VL, Brex PA, Chard DT, Thompson AJ. Interferon beta-1a in primary progressive multiple sclerosis: an exploratory, randomized, controlled trial.
Lovas G, Szilagyi N, Majtenyi K, Palkovits M, Komoly S. Axonal changes in chronic demyelinated cervical spinal cord plaques.
Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis.
Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination.
Marburg O. Die sogenannte ‘akute Multiple Sklerose’.
Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis.
Pelletier D, Nelson SJ, Oh J, Antel JP, Kita M, Zamvil SS, et al. MRI lesion volume heterogeneity in primary progressive multiple sclerosis in relation with axonal damage and brain atrophy.
Peterson JW, Bö L, Mörk S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions.
Pittock SJ, Mayr WT, McClelland RL, Jorgensen NW, Weigand SD, Noseworthy JH, et al. Disability profile of multiple sclerosis did not change over 10 years in a population-based prevalence cohort.
Prineas JW, Kwon EE, Cho ES, Sharer LR, Barnett MH, Oleszak EL, et al. Immunopathology of secondary-progressive multiple sclerosis.
Revesz T, Kidd D, Thompson AJ, Barnard RO, McDonald JW. A comparison of the pathology of of primary and secondary progressive multiple sclerosis.
Richert ND, Ostuni JL, Bash CN, Duyn JH, McFarland HF, Frank JA. Serial whole-brain magnetization transfer imaging in patients with relapsing-remitting multiple sclerosis at baseline and during treatment with interferon beta-1b.
Rocca MA, Iannucci G, Rovaris M, Comi G, Filippi M. Occult tissue damage in patients with primary progressive multiple sclerosis is independent of T2-visible lesions-a diffusion tensor MR study.
Rovaris M, Bozzali M, Santuccio G, Ghezzi A, Caputo D, Montanari E, et al. In vivo assessment of the brain and cervical cord pathology of patients with primary progressive multiple sclerosis.
Rovaris M, Bozzali M, Iannucci G, Ghezzi A, Caputo D, Montanari E, et al. Assessment of normal-appearing white and gray matter in patients with primary progressive multiple sclerosis: a diffusion-tensor magnetic resonance imaging study.
Sarchielli P, Presciutti O, Pelliccioli GP, Tarducci R, Gobbi G, Chiarini P, et al. Absolute quantification of brain metabolites by proton magnetic resonance spectroscopy in normal-appearing white matter of multiple sclerosis patients.
Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis.
Silver NC, Tofts PS, Symms MR, Barker GJ, Thompson AJ, Miller DH. Quantitative contrast-enhanced magnetic resonance imaging to evaluate blood-brain barrier integrity in multiple sclerosis: a preliminary study.
Thompson AJ, Kermode AG, Wicks D, MacManus DG, Kendall BE, Kingsley DP, et al. Major differences in the dynamics of primary and secondary progressive multiple sclerosis.
Thompson AJ, Polman CH, Miller DH, McDonald WI, Brochet B, Filippi M, et al. Primary progressive multiple sclerosis.
Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis.
Author notes
1Center for Brain Research, Medical University of Vienna and 2Department of Neurology, Municipial Hospital Lainz, Vienna, Austria, Departments of 3Neurology and 4Pathology, Mayo Clinic, Rochester, MN, USA, 5Department of Neuropathology, University of Göttingen, 6Institute for Multiple Sclerosis Research, University of Göttingen and Gemeinnützige Hertie-Stiftung and 7Department of Neuropathology, Zentralkrankenhaus Bremen-Ost, Germany


{kind=link}