


Abstract
Levetiracetam and its carboxylic metabolite (AcL) were tested for their potential inhibitory effect on 11 different drug metabolizing enzyme activities using human liver microsomes. The following specific assays were investigated: testosterone 6β-hydroxylation [cytochrome P-450 3A4 (CYP3A4)], coumarin hydroxylation (CYP2A6), (R)-warfarin hydroxylation (CYP1A2), (S)-mephenytoin hydroxylation (CYP2C19),p-nitrophenol hydroxylation (CYP2E1) tolbutamide hydroxylation (CYP2C9), dextromethorphan O-demethylation (CYP2D6), epoxide hydrolase and UDP-glucuronyltransferase (UGT) toward paracetamol (UGT1*6), ethinyloestradiol (UGT1*1),p-nitrophenol (UGT(pl 6.2)), and valproic acid. None of these activities were affected by levetiracetam or AcL added at concentrations up to 1 mM. Additionally, primary cultures of rat hepatocytes were used to assess a potential inducing effect of levetiracetam on CYPs. Phenobarbital (2 mM), β-naphtoflavone (40 μM), dexamethasone (1 μM), and phenytoin (up to 300 μM) were tested as positive controls. When added to cells for 48 h, all the positive controls increased 7-ethoxycoumarinO-deethylase activity demonstrating the inducibility of CYPs in the present culture conditions. By contrast, levetiracetam did not affect the activity up to 1 mM. The highest levetiracetam concentrations examined in the above in vitro studies are well in excess of those measured in the plasma of patients receiving therapeutic doses. It is thus concluded that levetiracetam is unlikely to produce pharmacokinetic interactions through inhibition of CYPs, UGTs, and epoxide hydrolase. Furthermore, based on the in vitro assays with rat hepatocytes, it could be speculated that levetiracetam does not act as a CYP inducer.
Levetiracetam ([(S)-α-ethyl-2-oxo-1-pyrrolidine-acetamide], Fig.1) is a new agent with an original spectrum of activity in animal models of seizures and epilepsy (Klitgaard et al., 1998). It has been shown of therapeutic benefit as add-on treatment in patients with refractory partial complex seizures under polytherapy. Coadministration with antiepileptic drugs (AEDs)1 (e.g., phenytoin, carbamazepine, and valproate) is therefore anticipated in a large number of patients for extended period of time. In humans, levetiracetam shows limited metabolism, with 66% of the dose excreted in urine as parent drug. Its major metabolic pathway involves the hydrolysis of the acetamide group to yield a carboxylic derivative (AcL; Fig. 1), which is mainly recovered in urine (24% of the dose)2. This reaction is tentatively assumed to be supported by amidase, an enzyme widely distributed in the organism. Although these findings indicate that levetiracetam is not significantly metabolized by either cytochrome P-450s (CYPs) or UDP-lucuronyltransferases (UGTs), inhibitory or inducing effects on these latter enzymes could not be ruled out.

Structures of levetiracetam and its major metabolite.
Approximately 25% of patients suffering from intractable epilepsy are polymedicated with several AEDs, and these patients may experience drug interactions. Most of the clinically relevant pharmacokinetic interactions with AEDs are related to their extensive hepatic biotransformation and their potential to inhibit and/or to induce liver drug metabolizing enzymes (Patsalos and Duncan, 1993; Mawer and Pleuvry, 1995; Mather and Levy, 1996). Much attention has therefore recently been focused on in vitro methodologies allowing prediction of interactions involving different AEDs (Food and Drug Administration, 1995; Levy, 1995). In the different complementary in vitro studies described in the present work, human liver microsomes were used to screen the ability of levetiracetam and AcL to inhibit selected marker activities for CYPs, UGTs, and epoxide hydrolase (EH). In addition, the potential of levetiracetam to induce CYPs was investigated using primary cultures of rat hepatocytes. Its inducing potency was compared with those of phenobarbital, β-naphtoflavone, dexamethasone, and phenytoin.
Materials and Methods
Chemicals and Reagents.
Levetiracetam and AcL were synthetized at UCB S.A. Pharma Sector. Progabide was kindly donated by Dr. Rovei (Synthelabo, Bagneux, France). Tolbutamide, warfarin, coumarin, 7-hydroxycoumarin, paracetamol, p-nitrophenol, [14C]-paracetamol, ethinyloestradiol, Brij 58, uridine 5′-diphosphoglucuronic acid (UDPGA), and neutral red were obtained from Sigma Chemical Co. (St. Louis, MO). Glucose 6-phosphate (G6P), glucose 6-phosphate dehydrogenase (G6PDH) (from yeast, grad I), NADPH, and NADP were purchased from Boehringer Mannheim GmbH Biochemica (Mannheim, Germany). Valproic acid was obtained from Aldrich (Milwaukee, WI). Dextromethorphan was purchased from ICN Biomedicals (Costa Mesa, CA). Hydroxytolbutamide was obtained from Ultrafine Chemicals (Manchester, England). 4′OH-Mephenytoin was obtained from Research Biochemicals International (Natick, MA). Testosterone, (±)-1-phenyl-1,2-ethanediol, and styrene oxide were obtained from Fluka Chemika (Buchs, Switzerland). 6β-Hydroxytestosterone was obtained from Steraloids Inc. (Wilton, NH). [14C]-UDPGA and [3H]ethinyloestradiol were purchased from Dupont de Nemours (Wilmington, DE). (R)-Warfarin was resolved from the racemate as described by West et al. (1961) and 6-hydroxy (R)-warfarin was synthesized according toHermodson et al. (1971). (S)-Mephenytoin was synthesized as described by Wienkers et al. (1996). Diazomethane was generated from Diazald (Aldrich) according to the manufacturers’s recommendations. Other chemicals were of the highest purity available.
Human Liver Microsomes.
Human liver specimens were obtained under strict ethical conditions from organ donors. Microsomes were prepared by differential centrifugation of liver homogenate as described elsewhere (Kremers et al., 1981; Thummel et al., 1993). The microsomal pellet (105,000g) was suspended at a final protein concentration of approximately 5 mg/ml in either 50 mM Tris (pH 7.4) or 100 mM phosphate buffer (pH 7.4) containing 1 mM EDTA. Microsomes were stored frozen at −70°C until subsequent analysis. Microsomal protein concentration was determined by the Lowry assay (Lowry et al., 1951) using bovine serum albumin as standard.
Microsomal CYP Marker Activities.
The assays were performed according to established methodologies. Briefly, testosterone 6β-hydroxylation (CYP3A4) (Arlotto et al., 1991), coumarin hydroxylation (CYP2A6) (Miles et al., 1990), warfarin 6-hydroxylation (CYP1A2) (Bush et al., 1983), (S)-mephenytoin 4′-hydroxylation (CYP2C19) (Goldstein et al., 1994), and p-nitrophenol hydroxylation (CYP2E1) (Tassaneeyakul et al., 1993) were assayed using a substrate concentration of 100, 50, 500, 20, and 40 μM, respectively, approximately 1 mg/ml of microsomal protein, and 1 mM NADPH. Tolbutamide hydroxylation (CYP2C9) (Ho and Moody, 1992) and dextromethorphan O-demethylation (CYP2D6) (Wu et al., 1993) were determined using a substrate concentration of 300 and 50 μM, respectively, 0.4 mg/ml of microsomal protein, and a NADPH-generating system (1 mM NADP, 6 mM G6P, and 0.4 U/ml G6PDH). Reaction was stopped after 30 min incubation at 37°C for CYP2D6, CYP1A2, and CYP2E1 marker activities. A 10-, 15-, 20-, and 25-min incubation time was used for CYP3A4, CYP2A6, CYP2C9, and CYP2C19 marker activities, respectively. As a rule, the selected marker substrate concentrations corresponded to approximately 2.5 times the Km for the corresponding CYP isoform.
Microsomal UGT Activities.
UGT activities toward [14C]paracetamol (UGT1*6), [3H]ethinyloestradiol (UGT1*1), andp-nitrophenol (UGT(pl6.2)) were determined as described previously (Burchell and Weatherill, 1981; Pacifici et al., 1988,Pacifici and Back, 1988). Briefly, [14C]paracetamol UGT activity was measured following a 40-min incubation at 37°C of 3 mg microsomal protein/ml with 500 μM substrate and 5 mM UDPGA. [3H]Ethinylestradiol UGT activity was measured following a 80-min incubation at 37°C of 0.7 mg microsomal protein/ml with 192 μM substrate and 5 mM UDPGA. p-Nitrophenol UGT activity was measured following a 20-min incubation at 37°C of 0.4 mg microsomal protein/ml with 500 μM substrate and 4 mM UDPGA. All the assays were performed in 100 mM Tris buffer (pH 7.4) containing 5 mM MgCl2. For all the assays, the microsomal samples were activated by a preceding 30-min incubation at 4°C with Triton X-100 (final detergent to protein ratio of 0.4 w/w). The exception was [14C]paracetamol UGT in which Brij 58 was used (final detergent to protein ratio of 0.4 w/w).
For valproic acid UGT assay, the microsomal sample was activated by a 20-min incubation at 4°C in 50 mM Tris buffer, pH 7.4, containing Brij 58 at detergent/protein ratio of 0.2 w/w. The microsomal sample (∼1 mg protein/ml) was then incubated at 37°C for 30 min in the presence of 1 mM valproic acid, 1 mM [14C]UDPGA (0.2 μCi/assay) in 50 mM Tris buffer (pH 7.4) containing 10 mM MgCl2. The final incubate volume was 0.5 ml. The reaction was stopped by addition of 250 μl ice-cold CH3CN. After centrifugation at 10,000gfor 10 min, a 50-μl aliquot of the supernatant was analyzed by reversed phase high-performance liquid chromatography using a computer-controlled Kontron 400 system (Kontron, Milan, Italy) consisting of a 420 pump, a 465 automatic sample injector, and a 425 gradient former. The system was fitted with a LB507A radioactivity monitor and a LB5035 scintillator pump (Berthold, Wildbad, Germany). Valproic acid and its glucuronide were resolved at room temperature on a Nucleosil 120-3C18 column (3 μm, 3 × 125 mm; Machery-Nagel, Düren, Germany), protected by a guard column (3 × 10 mm) of the same material. An isocratic elution with 10 mM ammonium acetate containing 0.1% diethylamine (pH 2.6) and CH3CN (80:20, v/v) was operated at 0.5 ml/min. The eluate was mixed with scintillation cocktail at a flow rate of 3 ml/min and passed through the radioactivity monitor fitted with a flow cell of 2 ml.
Microsomal EH Activity.
The microsomal sample was incubated at the final protein concentration of 50 μg/ml with 100 μM styrene oxide in 100 mM phosphate buffer (pH 7.4). After a 10-min incubation at 37°C, the reaction was stopped by addition of n-hexane. The sample was centrifuged at 1000g for 5 min and phenylethanediol formed was extracted from the aqueous phase and quantified by high-performance liquid chromatography according to Kerr et al. (1989).
In Vitro Inhibition of CYP, UGT, and EH Marker Activities.
In inhibition experiments, levetiracetam and AcL were added to the microsomal incubates just before initiation of the reaction. For all the assays, levetiracetam and AcL were added as stock solutions in 100 mM Tris buffer (pH 7.4), except for CYP1A2, CYP2C19, and CYP2E1 activities in which 100 mM phosphate buffer (pH 7.4) was used. An equivalent volume of vehicle was added to control incubates. Progabide was assayed as positive control in the EH inhibition study. In this last assay, progabide was added as a methanolic solution (1% v/v final concentration in the incubate). The incubation conditions for all the activities had been optimized for protein and time linearity. Furthermore, all the marker activities have been previously demonstrated to respond to specific chemical inhibitors. As a rule, inhibition assays were performed in three representative human liver microsomal samples.
Mean control activities in the microsomal samples used in the inhibition assays were for testosterone 6β-hydroxylation, 6.5 nmol/mg/min; tolbutamide hydroxylation, 0.35 nmol/mg/min; dextromethorphan O-demethylation, 0.33 nmol/mg/min; coumarin hydroxylation, 1.7 nmol/mg/min; (R)-warfarin hydroxylation, 55 pmol/mg/min; (S)-mephenytoin hydroxylation, 64 pmol/mg/min; p-nitrophenol hydroxylation, 1.8 nmol/mg/min; paracetamol UGT, 0.26 nmol/mg/min; ethinyloestradiol UGT, 65 pmol/mg/min; p-nitrophenol UGT, 23 nmol/mg/min; valproic acid UGT, 3.0 nmol/mg/min; and EH, 118 nmol/mg/min.
Rat Hepatocytes Isolation and Culture.
Hepatocytes were isolated from fasted male Sprague-Dawley OFA SPF rats (200–300 g) using a modification of Seglen’s two-step perfusion technique (Seglen, 1976). At isolation, cell viability as assessed by trypan blue exclusion was higher than 80%. Cells were suspended in Williams’ E medium supplemented with 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 10% v/v heat-inactivated fetal bovine serum and were seeded on 60 mm Nunc dishes (1 × 105 viable cells per cm2) previously coated with collagen S. Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2.
Hepatocyte Treatment and End Points Measurement.
Cells were allowed to attach for 3 h, at which time they were shifted to Williams’ E medium supplemented with 2 mMl-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 nM insulin, 10 nM dexamethasone, and the indicated xenobiotics. The medium was renewed after 24 h. At 48 h, cells were analyzed for 7-ethoxycoumarin O-deethylase (ECOD) activity and neutral red uptake. Phenobarbital and levetiracetam were added directly to culture medium. Stock solutions of phenytoin and β-naphtoflavone were prepared in dimethylsulfoxide, whereas ethanol was used for dexamethasone. Stock solutions were added to cultures at a final volume of 1% v/v. Control cells were incubated with the appropriate vehicles.
Determination of ECOD was performed on intact cells. Briefly, cultures were washed twice with phosphate buffered saline and then incubated at 37°C with Hank’s balanced solution containing 300 μM ethoxycoumarin. After a 20-min incubation at 37°C, cells were homogenized in the culture medium and frozen at −20°C until analysis. A sample of homogenate was saved for protein measurement using bovine serum albumin as the standard (Smith et al., 1985). A 400-μl homogenate sample was thawed, mixed with 600 μl of glucuronidase (1.4 mg/ml in 100 mM Na acetate buffer, pH 4.5, containing 100 mM NaCl), and incubated for 1 h at 37°C to allow hydrolysis of hydroxycoumarin conjugates. Hydroxycoumarin formed was extracted and measured using the fluorimetric method described byEdwards et al. (1984). Cells exposed to levetiracetam and phenytoin were assayed for neutral red uptake, as an indicator of cell toxicity (Zhang et al., 1990).
Results
Inhibition of CYP Marker Activities.
The potential in vitro inhibitory effect of levetiracetam andAcL on CYP3A4, CYP2C9, CYP2D6, CYP2A6, CYP1A2, CYP2C19, and CYP2E1 was quantitated by measuring their effect on specific marker activities, namely testosterone 6β-hydroxylation, tolbutamide hydroxylation, dextromethorphan O-demethylation, coumarin hydroxylation, (R)-warfarin hydroxylation, (S)-mephenytoin hydroxylation, and p-nitrophenol hydroxylation. At the final concentration of 1 to 1.25 mM, levetiracetam and AcL did not produce relevant inhibition of the investigated activities (≤11% inhibition).
Inhibition of UGT Marker Activities.
UGT activities toward paracetamol, ethinyloestradiol,p-nitrophenol, and valproic acid were not affected by either levetiracetam or AcL added at the final concentration of 1 mM (≤6% inhibition).
Inhibition of EH.
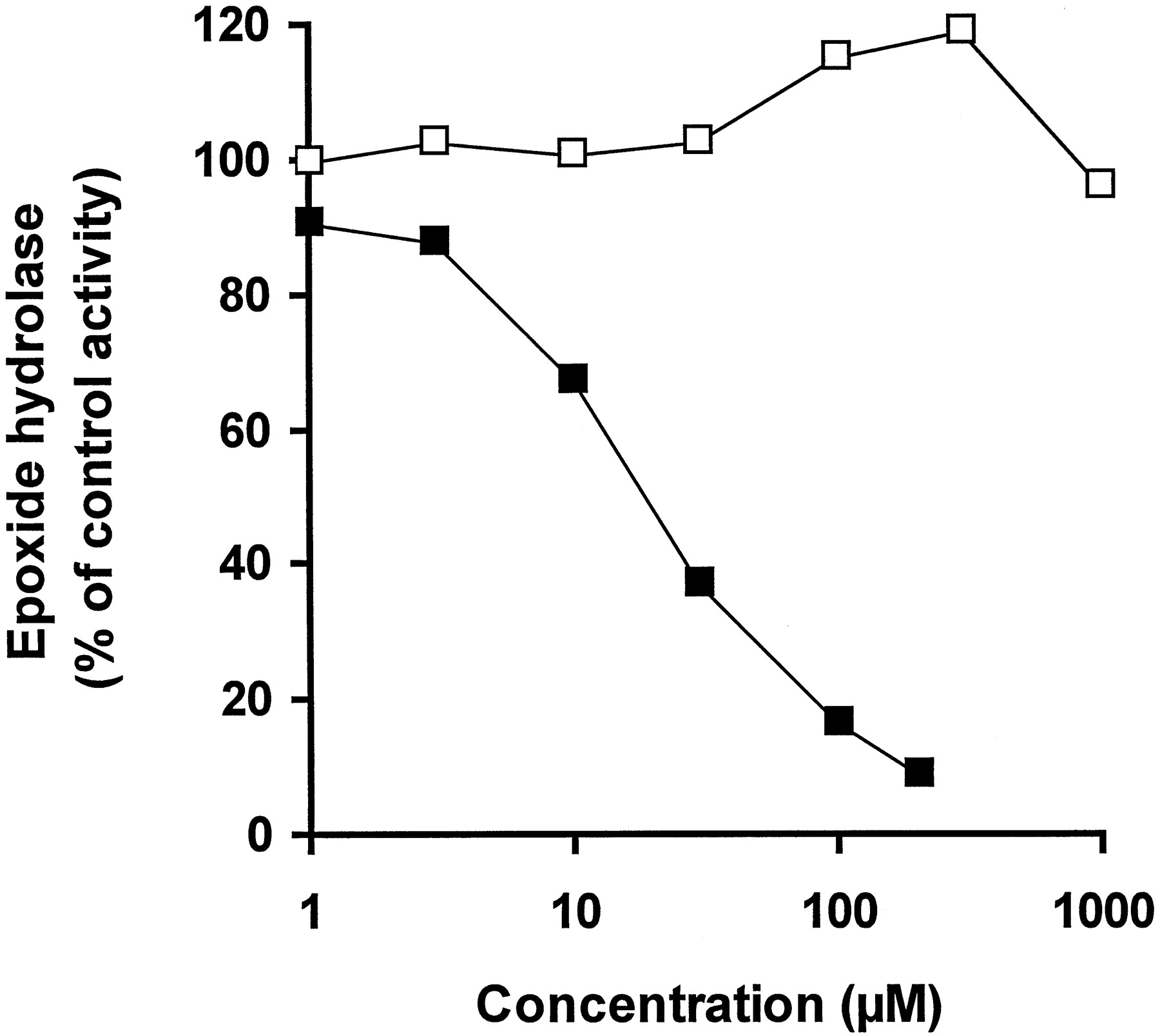
Up to the highest tested concentration of 1 mM, levetiracetam did not affect human liver microsomal EH activity. In contrast, progabide inhibited the activity with an IC50 of 19.7 μM (Fig. 2). At 1 mM, AcLremained without any effect (103% of the vehicle control value).

Effect of levetiracetam and progabide on EH.
EH activity was measured in a representative human liver microsomal sample with increasing concentrations of either levetiracetam (■) or progabide (▪). Activity is expressed as a percentage of control activity measured in the presence of vehicle.
Induction of CYP in Primary Cultures of Rat Hepatocytes.
A preliminary assay was conducted to examine the cytotoxic potential of levetiracetam and phenytoin when incubated with primary cultures of rat hepatocytes for 48 h. Levetiracetam and phenytoin did not modify neutral red uptake up to 1 mM and 300 μM, respectively (Fig.3).

Cytotoxicity of levetiracetam and phenytoin when incubated with primary cultures of rat hepatocytes.
Cells were exposed to levetiracetam (■) and phenytoin (▪) for 48 h before neutral red uptake measurement. Medium was renewed 24 h after treatment initiation. Activity is expressed as a percentage of control activity measured in the presence of vehicle. Results are mean of two independent cell preparations.
To determine the inducibility of CYPs in the present culture conditions, cells were exposed to noncytotoxic concentrations of well-characterized in vitro inducers, i.e., 2 mM phenobarbital, 40 μM β-naphtoflavone, and 1 μM dexamethasone. In agreement with literature data (Bars et al., 1989; Wortelboer et al., 1991), ECOD activity was induced 4.8-, 13.5-, and 3.6-fold by phenobarbital, β-naphtoflavone, and dexamethasone, respectively.
At 300 μM, phenytoin produced a 4-fold increase in ECOD activity, whereas levetiracetam remained without any significant effect up to 1 mM (Fig. 4).

Effect of levetiracetam on ECOD activity in primary cultures of rat hepatocytes.
Cells were exposed to xenobiotics for 48 h before ECOD activity measurement. Medium was renewed 24 h after treatment initiation. Results are expressed as fold increase above corresponding control cells (mean and S.D. of three independent cell preparations). Dexamethasone, phenobarbital, and β-naphtoflavone were evaluated in a separate experiment.
Discussion
The most important pharmacokinetic interactions encountered in epilepsy treatment are those in which the metabolism of some AEDs (e.g., phenytoin, carbamazepine, and valproic acid) is inhibited, precipitating their side effects. Alternatively, AEDs may induce enzymes involved in the metabolism of a number of drugs (e.g., corticoids, oral contraceptives, psychotropic drugs, oral coagulants, cardiovascular agents, and analgesics), thereby reducing their efficacy. AEDs inducing metabolism include carbamazepine, phenytoin, phenobarbital, and primidone. Levetiracetam is likely to be coadministered with other AEDs or drugs belonging to other therapeutic classes and hence, the risk for drug interaction must be considered.
In humans, levetiracetam is mainly excreted unchanged and its major metabolic pathway does not seem to involve either CYPs or UGTs. Thus, levetiracetam disposition is unlikely to be affected when concomitantly administered with inhibitors of these enzymes. However, this finding does not preclude an effect of levetiracetam on the metabolism of coadministered drugs. Indeed, there is a great deal of precedent in the literature for drugs affecting human metabolizing enzymes for which they are not substrates (for a review see Parkinson, 1996). For example, quinidine is a potent competitive inhibitor of CYP2D6, even though its disposition is largely determined by the CYP3A4. Similarly, phenytoin induces CYP3A4, whereas its biotransformation is supported by isoforms of the CYP2C subfamily.
Consequently, this work concentrated on the potential inhibitory effects of levetiracetam and its major metabolite, AcL, on major liver drug metabolizing (iso)enzymes. At the final concentration of 1 mM (i.e., 170 μg/ml), levetiracetam and AcL were without any effect on human liver microsomal marker activities for CYP3A4, CYP2A6, CYP1A2, CYP2C19, CYP2E1, CYP2C9, CYP2D6, UGT1*1, UGT1*6, and UGT(pI6.2). In addition, these compounds failed to interfere with EH and valproic acid glucuronidation. The highest levetiracetam concentration used in these assays is well in excess of those measured in the plasma of patients receiving therapeutic doses (i.e., 6 to 60 μg/ml; data on UCB files and Ratnaraj et al. 1996). Thus, levetiracetam is unlikely to produce clinically relevant interactions through inhibition of the above-mentioned (iso)enzymes.
Absence of inhibitory effect of levetiracetam on CYPs, UGTs, and EH is of major importance, because most of these enzymes are involved in AED metabolism (for a review see Patsalos and Duncan, 1993; Levy, 1995). Phenytoin is eliminated principally by hydroxylation top-hydroxyphenytoin, a reaction catalyzed primarily by CYP2C9 and CYP2C19. On the other hand, epoxidation of carbamazepine to carbamazepine-10,11-epoxide is supported by CYP3A4. In addition, this latter CYP isoform is also involved in the metabolism of clonazepam, trimethadione, and zonisamide. Ethosuximide, felbamate, stiripentol, oxcarbazepine, eterobarb, and tiagabine were demonstrated to be metabolized through CYP-dependent pathways, the CYP isoforms involved being not yet fully characterized. Three members of the CYP2 family, CYP2C9, CYP2A6, and CYP2C19, have been recently described as responsible for the terminal desaturation of valproic acid into 4-ene-valproic acid, a minor metabolite possibly involved in valproic acid-mediated hepatotoxicity (Sadeque et al., 1995). Because some of the metabolites contribute to toxic and, possibly, therapeutic effects, it has become clinically important to recognize factors that may alter valproic acid biotransformation (Pisani, 1992).
As mentioned above, carbamazepine is metabolized into carbamazepine-10,11-epoxide by a CYP-dependent pathway. This major pharmacologically active metabolite is further converted into a diol derivative by EH. There is now increasing evidence that interactions producing an elevation of serum epoxide concentrations may precipitate carbamazepine toxicity (Patsalos and Duncan, 1993). Progabide, valpromide, and, to a lesser extent, valproic acid were demonstrated to inhibit EH both in vitro and in vivo (Kerr et al., 1989; Kroetz et al., 1993). As a consequence, their coadministration with carbamazepine results in elevated plasma levels of carbamazepine-10,11-epoxide and associated signs of neurotoxicity (Bianchetti et al., 1987).
Valproic acid undergoes an extensive hepatic biotransformation. Its direct conjugation with d-glucuronic acid is quantitatively the most important route with the resulting β-glucuronide excreted in the urine (Cotariu and Zaidman, 1988). UGT enzymes also play a major role in the metabolic excretion of other AEDs (e.g., felbamate, stiripentol, and remacemide) (Patsalos and Duncan, 1993; Patsalos and Sander, 1994) as well as miscellaneous drugs (Burchell et al., 1995). In addition, UGTs are involved in the metabolism of endogenous compounds, such as bilirubin, short-chain fatty acids, retinoic acid, biliary acids, and steroid and thyroid hormones. It has been suggested that some AEDs may cause biochemical or clinical abnormalities by interacting with these endogenous metabolic pathways (Perucca, 1987).
The potential of levetiracetam to induce CYPs was investigated in primary cultures of rat hepatocytes using 7-ethoxycoumarinO-deethylase activity as the end point. In rats, this latter activity is catalyzed by multiple CYP isoenzymes of the CYP1A1, CYP2A, CYP2B, CYP2C, and CYP3A subfamilies, with CYP1A1 giving the highest activity (Edwards et al., 1984). Data indicate that the cultured hepatocytes were responsive to phenobarbital, β-naphtoflavone, and dexamethasone, as protype inducers of rat liver CYP2B1/2, CYP1A1/2, and CYP3A1/2, respectively. In the same experimental model, even at the highest concentration of 1 mM, levetiracetam failed to induce ECOD activity. Interestingly, phenytoin induced this CYP-supported activity (4-fold increase at 300 μM). Treatment of rats with this latter antiepileptic was demonstrated to induce isoforms of the CYP2B subfamily (Nims et al., 1994). In contrast, in vitro and in vivo data suggest that phenytoin is a CYP3A4 inducer in humans (Pichard et al., 1990; Fleishaker et al., 1995). There are other examples in which rodents differ from humans in their responsiveness to enzyme inducers. For instance, rifampin is a potent inducer in humans and rabbits but not in rats and mice (Parkinson, 1996). Thus, although reassuring, the data obtained with levetiracetam should be interpreted with caution. Obviously, primary cultures of human hepatocytes should represent a more relevant in vitro approach to evaluate its inducing potential.
In conclusion, using complementary in vitro assays, it was demonstrated that levetiracetam has no potential to produce clinically relevant pharmacokinetic drug interactions through inhibition of liver drug metabolizing enzymes. Of importance, levetiracetam does not interfere with the different metabolic pathways involved in phenytoin, carbamazepine, and valproic acid biotransformation. In addition, rat hepatocytes were used as initial screen for CYP induction by levetiracetam. Although not completely predictive of the clinical situation, the data obtained indicate that levetiracetam is unlikely to produce in vivo induction of CYPs. Lack of interaction may be an additional benefit when applying levetiracetam as an add-on treatment of refractory epilepsy.
Acknowledgments
We are grateful for the technical assistance of C. Derwa, L. Lerat, I. Leysen, F. Mériaux, and to P. Piette for assistance with in vitro incubations and enzymatic activities measurements.
Footnotes
-
Send reprint requests to: Jean-Marie Nicolas, Department of Product Safety and Metabolism, UCB S.A. Pharma Sector, Chemin du Foriest, B-1420 Braine-l’Alleud, Belgium.
-
This work was performed in part at the University of Washington and supported by UCB S.A. Pharma Sector. A portion of this work was presented at the XIth International Symposium on Microsomes and Drug Oxidations, Los Angeles, 1996, and at the European Symposium on Prediction of Drug Metabolism in Man, Liège, 1998.
-
↵2 Data on UCB files.
- Abbreviations used are::
- AcL
- carboxylic metabolite
- AED
- antiepileptic drug
- CYP
- cytochrome P-450
- ECOD
- 7-ethoxycoumarin O-deethylase
- EH
- epoxide hydrolase
- G6P
- glucose 6-phosphate
- G6PDH
- glucose 6-phosphate dehydrogenase
- UDPGA
- uridine 5′-diphosphoglucuronic acid
- UGT
- UDP-glucuronyltransferase
- Received May 14, 1998.
- Accepted October 27, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}