Article Text

Abstract
Cerebral amyloid angiopathy related inflammation (CAA-I), previously described under various names, is a treatable encephalopathy usually occurring in older adults. Here, three patients are described with histopathologically confirmed CAA-I, and summarised data from the published literature are presented. CAA-I has a characteristic combination of clinical and radiological features. Definite diagnosis requires brain and leptomeningeal biopsy. A favourable response to immunosuppressive therapy is common and treatment without brain biopsy may be considered in selected patients. Diagnostic criteria for CAA-I are proposed.
- Cerebrovascular disease
Statistics from Altmetric.com
Introduction
Cerebral amyloid angiopathy (CAA) is characterised by β amyloid deposition in the walls of small to medium sized arteries, and less frequently veins and capillaries, of the leptomeninges and brain.1 2 CAA can occur sporadically or rarely as a hereditary syndrome.3 The pathogenesis remains uncertain but it is likely that amyloid is produced locally in the brain and accumulates as a result of reduced clearance.4–6
CAA is found in between 23% and 57% of an asymptomatic elderly population on histopathology examination.7 CAA is also found in a number of disease states, most commonly dementia and intracerebral haemorrhage (ICH). In a review of four population studies with histopathological data, 55–59% of patients with dementia had evidence of CAA compared with 7–24% of patients without dementia.8 CAA associated ICH accounts for 10–34% of ICH and typically occurs in patients in their eighth decade or older. CAA associated ICH is more likely to be lobar in location, is often multifocal and tends to recur more frequently than hypertensive haemorrhage. CAA is also associated with transient neurological symptoms, particularly in older patients, who may present with multiple, stereotyped episodes of predominantly sensory symptoms. These transient symptoms are presumed to be focal seizures and the episodes often respond to antiepileptic medication.
A less common manifestation is CAA related inflammation (CAA-I). CAA-I presents with acute or subacute onset of headaches, cognitive and behavioural changes, seizures and focal neurological deficits. In addition to CAA-I, this condition has been labelled primary angiitis of the CNS associated with CAA; amyloid angiopathy and granulomatous angiitis of the CNS; cerebral amyloid inflammatory vasculopathy; cerebral amyloid angiitis; cerebral amyloid angiopathy associated with giant cell arteritis; and amyloid β related angiitis.9–15 All of these names emphasise the association between cerebral amyloid angiopathy and inflammation of blood vessels. We prefer the label CAA-I as perivascular inflammation may also be found.
We report three patients with pathologically confirmed sporadic CAA-I who presented between 2001 and 2007. Patients were included if they presented with encephalopathy and had histopathological evidence of perivascular or intramural vascular inflammation and deposition of amyloid in blood vessels in those parts of the cerebral cortex and leptomeninges affected by the inflammatory changes. The findings from 69 previously published cases are summarised. We propose diagnostic criteria for CAA-I.
Case reports
Patient No 1
An 83-year-old woman presented with a first generalised tonic–clonic seizure. Her past history included cognitive impairment for 2 years, depression, rheumatoid arthritis for 13 years, giant cell arteritis diagnosed 15 years earlier and basal cell carcinoma over the left temple for which she had received radiotherapy 6 months earlier. She was taking methotrexate 15 mg weekly and prednisone 3 mg daily for arthritis. Mini-Mental State Examination score was 18/30. The examination was otherwise unremarkable.
T2 weighted and fluid attenuation inversion recovery (FLAIR) MRI sequences showed asymmetric, confluent and patchy hyperintensities in both cerebral hemispheres (figure 1A). Susceptibility weighted sequences were not performed. Blood tests were unremarkable. CSF was abnormal with raised protein (1.05 g/l; normal 0.15–0.45) but normal glucose content and cell count. PCR for JC virus was negative. Phenytoin was commenced, prednisone was continued at the same maintenance dose and methotrexate was stopped.

An 83-year-old woman presenting with a first generalised tonic–clonic seizure and a Mini-Mental State Examination score of 18/30. (A) MRI fluid attenuation inversion recovery sequence demonstrating asymmetric, bilateral white matter hyperintensity. (B) High power view of the histology demonstrating transmural inflammation of a cortical blood vessel with red blood cells in the lumen (arrowhead) and a multinucleated giant cell (long arrow). (C) Perivascular inflammation, amyloid deposition in artery (thin arrow), epithelioid cell (arrowhead) and multinucleated giant cell (thick arrow) (C).
Over the next 2 weeks she developed progressive loss of consciousness. Repeat MRI showed progression of the left frontal changes on T2 weighted sequences. A repeat CSF examination found 9×106/l white cells (46% lymphocytes, 40% monocytes, 10% polymorphs, 4% degenerate cells), protein 2.95 g/l and normal glucose. She continued to deteriorate and died 1 month after presentation.
A postmortem examination of the brain and leptomeninges showed deposition of amyloid in leptomeningeal and cortical vessel walls with a ‘double lumen’ in some arteries and changes consistent with granulomatous vasculitis. Multinucleate giant cells were present, mainly confined to the perivascular space (figure 1B,C). In a few arteries the inflammation penetrated through the media. There was a moderate number of neuritic plaques and neurofibrillary tangles in the frontal cortex, suggestive of mild Alzheimer's disease. Quantification of the number of plaques and tangles was not performed.
Patient No 2
A woman in her forties presented to another hospital with a 1 day history of severe headache followed by mild left hemiparesis and sensory loss. Her past history included type II diabetes mellitus, hyperlipidaemia and morbid obesity. CT showed a right posterior parietal hypodensity consistent with subacute cerebral infarction. She was unable to have MRI because of large body habitus. She was treated as having right middle cerebral artery territory infarction and was started on vascular secondary prevention and discharged.
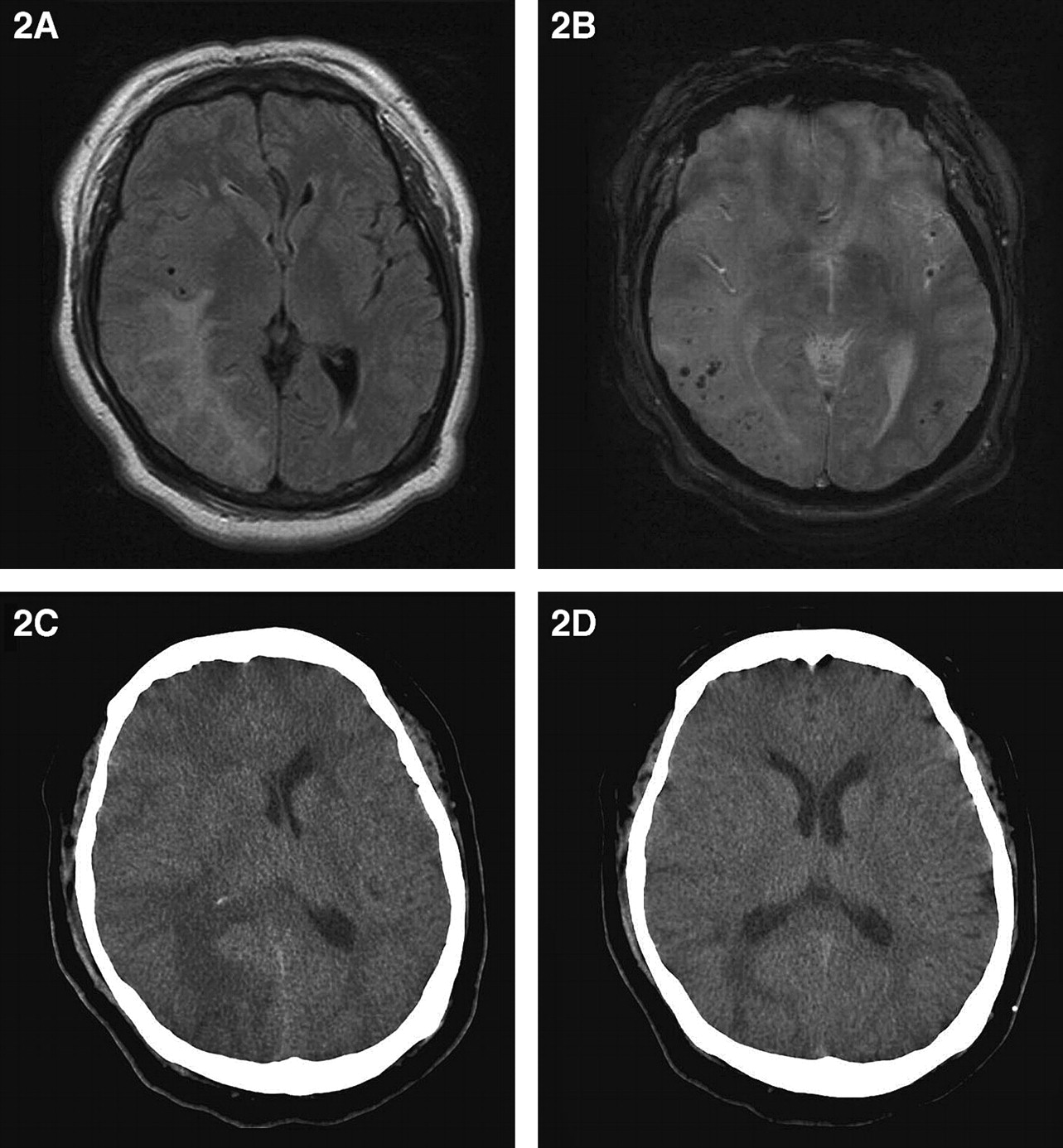
She presented 5 months later with a 2 day history of headache. Physical examination and routine blood tests were unremarkable. CSF analysis showed normal cell count and glucose but protein was elevated at 1.29 g/l. She had lost weight and was able to have MRI (figure 2A,B). T2 weighted and FLAIR sequences showed a right parieto-occipital lesion and susceptibility weighted sequences showed multiple microhaemorrhages throughout both cerebral hemispheres. A right parietal brain and dural biopsy showed amyloid deposition in the walls of the meningeal and cortical blood vessels. Smaller arteries had perivascular epithelioid cells, macrophages and occasional aggregates of lymphocytes. Larger arteries had transmural inflammation and poorly defined granulomatous inflammation.

{kind=link}
{kind=link}
A 46-year-old woman who initially presented with a 1 day history of severe headache followed by mild left hemiparesis and sensory loss. (A) MRI fluid attenuation inversion recovery sequence demonstrating large confluent right parieto-occiptal hyperintensity. (B) MRI susceptibility weighted sequence demonstrating multiple bilateral microhaemorrhages. (C) CT brain scan demonstrating extensive right cerebral hemisphere oedema with midline shift and a tiny deep focus haemorrhage. (D) CT brain after 6 months demonstrating marked reduction of cerebral oedema while on maintenance dexamethasone therapy.
She was treated with dexamethasone 1 mg twice daily and the headache resolved. The patient declined any other immunosuppressive therapy. She then stopped dexamethasone of her own accord and failed to attend for outpatient follow-up.
She re-presented 5 months later with recurrence of headache. The neurological examination was normal. However, a CT brain scan showed recent haemorrhage into the persisting right parieto-occipital lesion associated with oedema and significant midline shift (figure 2C). Dexamethasone was restarted leading to prompt symptomatic relief. The right parieto-occipital lesion was still present on a CT 7 months later although there was less associated oedema (figure 2D). She was prescribed dexamethasone 1 mg twice daily.
Patient No 3
This 72-year-old man presented with a generalised tonic–clonic seizure. His past history included multiple myeloma treated with melphalan and prednisone 2 years earlier. The myeloma was in remission. He recalled an episode of transient left-sided weakness 3 months earlier for which he did not seek medical attention. On examination he had left-sided neglect, a left homonymous hemianopia and reduced spontaneous movement in the left limbs without weakness.
T2 weighted and FLAIR MRI sequences showed white matter hyperintense lesions that were extensive and confluent in the right cerebral hemisphere and patchy in the left cerebral hemisphere. Susceptibility weighted sequences were not performed. Blood tests and a bone marrow examination were unremarkable. A brain and leptomeningeal biopsy showed prominent amyloid deposition in the walls of the leptomeningeal and cortical blood vessels. One meningeal artery showed foamy macrophages and transmural infiltration by epithelioid cells. There was a single area with granulomatous change. A few other arteries showed ill defined inflammatory cells in the perivascular space.
He was treated with methylprednisone and pulsed high dose intravenous cyclophosphamide. There was only minor clinical improvement and he tolerated the therapy poorly. Cyclophosphamide was stopped and prednisone continued at 20 mg/day. He died 14 months after initial presentation. No postmortem examination was performed.
Review of reported CAA-I cases
Sixty-nine patients with pathologically confirmed CAA-I have been reported in the literature.1 10 11 13–42 Information about demographic data, clinical presentation, investigations, treatment and outcome of these 69 patients have been extracted from the reports and, together with the three patients reported here, are summarised in table 1. Thirty-nine (54%) were men and mean age was 63 years (median 69, range 42–84 years). Seventeen (24%) were less than 60 years of age and seven (10%) were less than 50 years.
Cerebral amyloid angiopathy related inflammation summary: current three patients and those reported in the literature
Clinical features
Two of 14 patients reported by Kinnecom et al were excluded from analysis of clinical features because of lack of data.17 Patients with CAA-I presented acutely or subacutely. Over the course of the illness, cognitive and behavioural changes were seen in 53 of 70 (76%) patients where this information was reported. Features included poor memory, impaired concentration, dementia, personality and behavioural changes, confusion and altered level of consciousness. Headache occurred in 29 patients (41%) and seizures in 22 (31%). Patients rarely had constitutional symptoms and only five (7%) patients had fever.16 24 26 30 33 Focal neurological signs were seen in 32 patients (46%) and included dysphasia in 18 (26%), visual field defects in 12 (17%), monoparesis or hemiparesis in 12 (17%), cerebellar ataxia in six (9%) and hemineglect in five (7%). Some patients had rapid changes in laterality of the focal neurological features during the course of the illness.33 Seventy-nine per cent of the patients had two or more of the following: headache, seizures, mental status or behavioural change, and focal neurological deficit. Three patients (including patient No 2 described here) had transient ischaemia attack or stroke-like onset.16 33 In general, CAA-I was not associated with an underlying systemic inflammatory disorder.15
Investigations
Inflammatory markers including erythrocyte sedimentation rate, C reactive protein and plasma viscosity were elevated in 13 of 40 patients (33%) where this information was reported. Ten of 14 patients (71%) were positive for the APOE ε4/ε4 genotype where this was looked for.17 39 CSF showed pleocytosis in 19 of 42 patients (45%) and elevated protein in 30 of 42 (71%; range 0.5–5.73 g/l). An eosinophilic pleocytosis was sometimes observed.10 CSF oligoclonal bands were not found in the four cases where this was reported.10 12 27 41 CSF opening pressure was greater than 50 cmH2O in some patients.34 Where electroencephalography was reported, 18 of 19 patients (95%) had non-specific changes with either generalised or focal slowing, and one showed epileptiform discharges.
Fifty-five of 70 (79%) patients had MRI reported. Abnormalities were seen in all but two scans. Typical findings included patchy or confluent, asymmetric T2 weighted or FLAIR white matter hyperintensities. The appearances resembled a tumour in some cases.12 The use of contrast was not systematically reported but 17 patients had patchy contrast enhancement in the leptomeninges or parenchymal lesions. Susceptibility weighted imaging sequences were abnormal in 10 out of 11 (91%) patients where these sequences were reported (including one of our patients). Microhaemorrhages were present in nine patients and subcortical haemorrhage in one patient.
Eighteen patients underwent catheter cerebral angiography. This was abnormal in four patients (22%) with subtle bilateral vascular narrowing affecting small branch vessels of the middle and anterior cerebral arteries (one patient), alternating areas of vasoconstriction and normal calibre involving large and medium sized cerebral arteries (n=1), hypoplasia of the anterior cerebral artery (n=1) and eccentric narrowing of a few medium sized vessels (n=1). Eight patients had normal MR angiography including one with abnormal catheter angiography.
All 72 patients had a tissue diagnosis of CAA-I; 56 had a brain biopsy, 14 had a postmortem brain examination and two had both. All patients had vascular amyloid deposition accompanied by perivascular, intramural and/or transmural inflammatory changes, with or without granuloma formation. Ten patients (14%) had only perivascular inflammation, with or without multinucleate giant cells.9 21 26 36 Three patients had additional inflammatory changes affecting blood vessels harbouring little or no amyloid.13
Therapy
Fifty-three of 70 patients (76%) were treated with immunosuppressive therapy, although in patient No 1 reported here this comprised long term weekly methotrexate and low dose prednisone. Fifty-two of 53 patients were treated with corticosteroids. Nineteen received additional immunosuppressive therapy including cyclophosphamide (n=19), methotrexate (n=2) or mycophenolate mofetil (n=2).28 41 One patient was treated with cyclophosphamide alone.25
Thirty-eight of 53 treated patients (72%) showed clinical improvement, ranging from a transient response with subsequent deterioration to functionally relevant remission.16 Twenty (38%) had a continued response to therapy for up to 112 months. A response was generally seen 1–3 weeks after treatment was commenced.16 17 Twelve patients (23%) who deteriorated on treatment or relapsed after treatment withdrawal responded to reinstitution of treatment or increased dose of immunsuppressive therapy. Where follow-up MRI was reported, 24 of 32 treated patients (75%) had improvement of white matter lesions. One patient developed new intracerebral haemorrhage on therapy.17
Prognosis
Follow-up was reported in 70 patients and ranged from 3 days to 13 years. Twenty-three patients (33%) had died. Where the details were available, 16 of 33 survivors (48%) were asymptomatic or only mildly disabled.
Discussion
This case series and review of reported patients has summarised the typical presentation, clinical features, results of investigations, treatment and prognosis of CAA-I. CAA-I is a rare condition thought to be due to an inflammatory response to β amyloid (Aβ) in the walls of blood vessels. Aβ is produced locally by the action of β and γ secretase on the transmembrane protein amyloid precursor protein. There are three main mechanisms by which Aβ amyloid is cleared from the brain: degradation by metallopeptidases; direct absorption into the bloodstream via low density lipoprotein receptor related protein or P-glycoprotein on the luminal aspect of endothelial cells; and drainage with other solutes and interstitial fluid along capillary and artery walls.4–6 Impairment in one or more of these clearance mechanisms may result in the accumulation of Aβ in the walls of small and medium sized leptomeningeal and cortical blood vessels.
Several observations suggest that CAA-I is not simply the chance association of a rare disorder, primary CNS vasculitis (PCNSV), with a common one, CAA. Firstly, the radiological features found in CAA-I, particularly the confluent areas of T2 hyperintensity without evidence of infarction, would be unusual in PCNSV. Secondly, there is striking co-localisation of the vascular amyloid deposits with the inflammatory response. Thirdly, evidence that CAA-I is caused by an inflammatory response to Aβ deposition in the walls of blood vessels came from a clinical trial in which patients received vaccinations to the Aβ42 form of Aβ as a potential therapy for Alzheimer's disease.43–45 The trial was stopped early when 18 of 298 patients (6%) developed a subacute meningoencephalitis. These 18 patients had an acute or subacute neurological decline similar to the clinical features described in CAA-I. MRI findings were variable but most patients had posterior cerebral cortical and cerebellar lesions. Two of the patients have had postmortem examinations which showed inflammatory changes surrounding Aβ containing blood vessels.44 45
Clinical features and course
CAA-I affects men and women equally. Mean age of onset is in the seventh decade but cases (including one of our patients) occur in the fifth decade. Patients with CAA-I tend to be younger than those with CAA (mean age 67 years compared with 76 years) but older than those with PCNSV (mean age 45 years).15 16 The onset of symptoms is acute or subacute and includes a combination of headache, cognitive decline, seizures, encephalopathy and focal deficits. A small proportion of patients may present with a transient ischaemia attack or stroke-like event.
The clinical course of CAA-I is varied. There is usually a favourable response to immunosuppressive therapy. One patient with fluctuating aphasia and biopsy proven CAA-I responded to a 2 day course of corticosteroid therapy and remained neurologically normal 2 years later.36 However, patients may have a fluctuating course or relentless progression and die despite immunosuppressive therapy.16 34
Differential diagnosis
The major differential diagnoses include infections (particularly progressive multifocal leucoencephalopathy), neurosarcoidosis, other immune related conditions such as acute disseminated encephalomyelitis46 and malignant processes, such as primary CNS lymphoma,33 carcinomatous meningitis and gliomatosis cerebri. Posterior reversible encephalopathy syndrome also must be considered but the elevated blood pressure and typical precipitants usually associated with this condition help to clarify the diagnosis. Multiple microhaemorrhages may also be seen in hypertensive vasculopathy but the microhaemorrhages are usually found in the thalamus, brainstem, cerebellum and basal ganglia rather than in a lobar position.47
Investigation
An increase in inflammatory markers is frequently seen in CAA-I but is non-specific. The CSF is commonly abnormal with a mild to moderate lymphocytic pleocytosis, or elevation in protein levels, or both. CSF examination is required to help exclude infectious diseases such as tuberculosis, viral encephalitis, fungal infections, partially treated bacterial infections and neoplastic processes. MRI shows asymmetric or symmetric, confluent or patchy T2 weighted or FLAIR hyperintense lesions with or without oedema, and in some patients leptomeningeal or parenchymal enhancement. The MRI appearances may be mistaken for a malignant lesion. Multiple cortical and subcortical microhaemorrhages on susceptibility weighted sequences are frequently seen and help to differentiate CAA-I from PCNSV and posterior reversible encephalopathy syndrome. There also may be evidence of old lobar ICHs. Cerebral angiography is usually normal. Electroencephalography shows non-specific focal or generalised slowing or epileptiform changes.
A definite diagnosis of CAA-I can only be made with cerebral and leptomeningeal biopsy. Tissue samples should be taken from a region which is abnormal on brain imaging. However, in some patients with typical clinical and radiological presentations, no inflammatory changes are seen, possibly due to sampling error.13 This is similar to the results of brain biopsy in PCNSV and probably reflects the patchy nature of the pathological processes.13 48–50
Diagnostic criteria
Kinnecom et al suggested that a diagnosis of probable CAA-I may be made on the basis of typical clinical and radiological findings without requiring a biopsy.17 We agree and propose diagnostic criteria for definite and probable CAA-I (box 1). These criteria were based on the clinical and imaging findings in the 72 patients reported with CAA-I and consideration of how this information should be applied in clinical practice. Probable CAA-I requires presentation with a variable combination of acute or subacute onset headache, mental status or behavioural change, focal neurological deficits and seizures. MRI should show patchy or confluent T2 weighted or FLAIR lesions with or without mass effect. In some patients there may be patchy contrast enhancement of the leptomeninges and brain parenchyma but diffuse enhancement suggests an alternative diagnosis. Multiple lobar microhaemorrhages and recent or past lobar intracerebral haemorrhages should also be seen. Blood tests, CSF examination and other investigations have a limited role in making a diagnosis of CAA-I but are required to exclude infectious and malignant conditions. A diagnosis of definite CAA-I requires these features plus tissue evidence of amyloid associated perivascular, transmural or intramural vascular inflammation in the brain or leptomeninges.
Box 1 Proposed diagnostic criteria for cerebral amyloid angiopathy related inflammation (CAA-I)
Probable CAA-I
All of the following:
Acute or subacute onset of symptoms.
40 years of age or older.
At least one of the following clinical features: headache, mental status or behavioural change, focal neurological signs and seizures.
MRI shows patchy or confluent T2 or fluid attenuation inversion recovery hyperintensity which is:
usually asymmetric
with or without mass effect
with or without leptomeningeal or parenchymal enhancement.
Evidence of pre-existing CAA on susceptibility weighted MRI sequences:
multiple cortical and subcortical haemorrhages or microhaemorrhages and/or
recent or past lobar haemorrhage
absence of neoplastic, infectious or other cause.
Definite CAA-I
All of the above plus histopathological confirmation with:
perivascular, transmural and/or intramural inflammation
amyloid deposition within vessels of affected area in the cortex and leptomeninges.
Treatment
Most patients with CAA-I show at least a partial response to high dose corticosteroids or other immunosuppressive agents. Clinical improvement is commonly associated with a radiological response. It is difficult to make a general recommendation on the management of individual patients but we believe it is reasonable to start therapy with high dose corticosteroids. Once clinical and radiological improvement is seen, consideration can be given to introducing further immunosuppressive therapy with pulsed cyclophosphamide, methotrexate or mycophenolate mofetil. Most experience has been with cyclophosphamide. However, the age and clinical state of the patient should be taken into account with cyclophosphamide reserved for younger more robust patients. Patients who respond to treatment tend to do so within the first few weeks. The optimal duration of therapy remains to be determined but should be based on the clinical and radiological responses. Relapse may occur with reduction or cessation of immunotherapy.
Treatment without biopsy
It may be reasonable to start a trial of immunosuppressive therapy without brain biopsy in selected patients with probable CAA-I.51 The disadvantage of this approach is ongoing diagnostic uncertainty. Concurrent antibiotic cover may be considered if an infective cause cannot be immediately excluded. In conditions such as primary brain malignancy, neurosarcoidosis and PCNSV, empiric management with corticosteroid therapy will not harm the patient in the short term. Brain biopsy should be reconsidered in patients with probable CAA-I who fail to respond to empiric high dose corticosteroid therapy within 3 weeks.
Conclusion
Although rare and under-recognised in the past, CAA-I has a characteristic presentation. We propose diagnostic criteria that could be viewed as a starting point for future consensus meetings. A diagnosis of probable CAA-I can be made if a patient 40 years or older presents with typical clinical and radiological features. Histopathological examination is required for a definite diagnosis of CAA-I. However, once infectious and other causes are thought unlikely, it is reasonable to treat selected patients with probable CAA-I with immunosuppressive therapy without brain biopsy.
References
Footnotes
Competing interests None.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.