Article Text

Abstract
OBJECTIVES To perform a comprehensive survey of myasthenia gravis in the county of Cambridgeshire, England, establishing contemporary epidemiological data.
METHODS Cases were ascertained from multiple sources. Prevalent patients were visited and assessed by means of a standardised questionnaire and examination complemented by review of medical case notes.
RESULTS One hundred cases were identified in a population of 684 000 (prevalence 15 per 100 000 population, 95% confidence intervals (95% CIs) 12–18). Thirty eight new diagnoses were made over a five year period providing an incidence of 1.1/100 000 population/year. The sex ratio was 2:1 F:M. After a mean follow up of 11.7 years, symptomatic disease was still restricted to ocular muscles in 25 patients. Thirty four of 100 patients underwent thymectomy a mean of 0.8 years after presentation, and a thymoma was present in 12. Highest remission rates were seen in patients presenting with generalised disease who underwent thymectomy but did not have a thymoma (27%). Cosegregation of an additional autoimmune disease occurred in 27 patients and in 24/49 (49%) women with onset<50 years of age.
CONCLUSIONS This, the second highest reported prevalence for myasthenia, is likely to be the result of optimum case ascertainment, increased disease duration, application of complex diagnostic tests, and the impact of an aging population leading to a relative increase in the prevalence of ocular myasthenia.
- myasthenia gravis
- prevalence
- incidence
Statistics from Altmetric.com
Although generally considered to be one of the rarer neurological diseases, myasthenia gravis is important because of advances made in understanding its immunological basis and the potential for treatment. Epidemiological studies which take into account advances in laboratory investigations allow the changing patterns of disease to be determined, provide information for health service planning, and allow the testing of aetiological hypotheses. These show a trend of increasing prevalence1 with relatively stable incidence which is interpreted as reflecting the impact of effective treatment and improved diagnostic methods. Wide fluctuations between geographical regions in morbidity statistics are seen in more recent examinations of disease frequency, and seem unlikely to be the result of these factors or study methodology alone.
Accurate epidemiological analysis of low frequency diseases requires a large population to identify significant numbers of affected people to disclose patterns of clinical phenotype. As a result, population based studies are scarce and most clinical reports on myasthenia gravis rely on consecutive case series culled from specialist neurology clinics which may not be entirely representative of the at risk population. We have applied an established procedure for epidemiological studies of neurological disease to survey a large population for myasthenia gravis and to examine patterns of clinical practice and disease phenotype.
Patients and methods
STUDY AREA
Cambridgeshire county lies within the East Anglian region of England. Neurological outpatient facilities are provided from district general hospitals in Peterborough and Huntingdon, which lie within the county boundaries; further outpatient facilities are available in Bury St Edmunds, Bedford, and Kings Lynn, which lie in adjoining counties. Each hospital is served by at least one consultant neurologist and their staff, and all inpatient investigation is performed at a single regional neurology unit in Cambridge, where records are held for all patients seen since 1965. The county and adjoining area are served by 426 general practitioners operating from 154 surgeries. Population figures for the study area are published by the Cambridgeshire County Council Population Research Group, and report a 1995 mid-year estimate of 684 000.2
METHODS
A provisional register was created from eight sources. Departmental notes were examined to identify people seen since 1965 in whom a diagnosis of myasthenia had been made; this included records from neurophysiological examinations. Seven additional sources were used to identify patients with myasthenia gravis. All general practitioners within the Cambridgeshire, Huntingdon, and Peterborough County Districts and adjoining areas were asked to provide names, addresses, and dates of birth of affected patients registered with the practice and permission was sought to approach these patients. Reminders were sent at two 3-monthly intervals to non-responding practitioners. The local branch secretary of the Myasthenia Gravis Society was asked to provide a list of affected members once permission had been obtained from those concerned. The local thoracic surgery service provided names of patients who had undergone thymectomy for myasthenia gravis and similar requests were made to the paediatric neurologists and ophthalmologists. A list of requests for acetylcholine receptor antibodies since 1980 was obtained from a computerised record system held in the department of immunology and these patients’ case notes were reviewed. Finally, computerised hospital databases provided a list of those patients admitted with a primary or secondary diagnosis of myasthenia gravis to Peterborough, Hinchingbrooke, Kings Lynn, or Addenbrooke’s Hospitals after 1985. Patients were considered prevalent if they were alive and normally resident in the area on 1 July 1997, and incident when the diagnosis of myasthenia had been made while they were resident in the study area.
After establishing that the patient was aware of the diagnosis, and with consent from the general practitioner, attempts were made to contact all patients on the provisional register to confirm residence within the study area and to collect clinical data by examination and a standardised questionnaire complemented by review of hospital or primary case notes. The diagnosis of myasthenia was based on three or more of the following: (1) typical history; (2) clinical evidence of fatiguability with recovery on rest; (3) clinical response to anticholinesterase administration; (4) detection of acetylcholine receptor antibodies; (5) decrement on electrical activity on repetitive stimulation; (6) exclusion of alternative relevant diagnoses. Disease classification was according to modified Osserman group definitions.3 New diagnoses of myasthenia made by Cambridge based neurologists for the study area were recorded prospectively on a central register from 1992.
Results
CASE ASCERTAINMENT
The provisional register contained 205 patients from all sources. Many were identified from more than one source (table 1). One hundred and fifty one (98%) of the 154 general practioner practices responded to the request for information. Ninety seven (47%) patients on the provisional register had either died, moved away from the area, were duplicate referrals, or an alternative diagnosis had been established, and these were all excluded. After further clinical evaluation based on personal interview or review of existing medical records, the diagnosis could not be confirmed in a further eight (4%) patients: alternate diagnoses comprised mitochondrial cytopathy (n=3), eyelid apraxia (n=1), idiopathic bulbar palsy (n=1), and focal dystonia (n=1); in a further two patients there was insufficient evidence to support the diagnosis of myasthenia even though no alternative seemed more likely.
Recruitment sources for 100 prevalent patients
The final register therefore comprised 100 patients with myasthenia gravis, alive and prevalent within the study area on 1 July 1997. The largest source for ascertainment was from general practitioner referrals (table 1), which also provide the largest number of patients identified from a single source (42, 42%; see table 1). Sixty one (61%) of the 100 prevalent patients were examined and interviewed and the remainder assessed on the basis of recent and comprehensive departmental notes.
PREVALENCE
The prevalence of myasthenia gravis in Cambridgeshire on 1 July 1997 was 100/684 000 population (15/100 000 population; 95% CI 12–18). Table 2 shows the age and sex specific prevalence of myasthenia gravis in Cambridgeshire, which varies from 0/100 000 in the 0–10 year age group to 63/100 000 population (males 86, females 53) in the 80+ age group. The sex ratio was 2:1 F:M. The mean age of prevalent patients was 59.7 (males 68.2, females 55.5, range 16–89) years; mean age at onset was 46.0 (males 59.0 females 39.4) years, mean age at presentation was 47.5 (males 60.8, females 40.7) years, and mean age at diagnosis 47.8 (females 41.1, males 70.0) years. Fifty six (56%) were aged 50 years or younger at diagnosis (males 7/33, 24%; females 49/67, 73%). A bimodal pattern of age at onset was seen in females peaking at 21–50 and 71–80 years together with a single peak in males at 61–70 (figure).
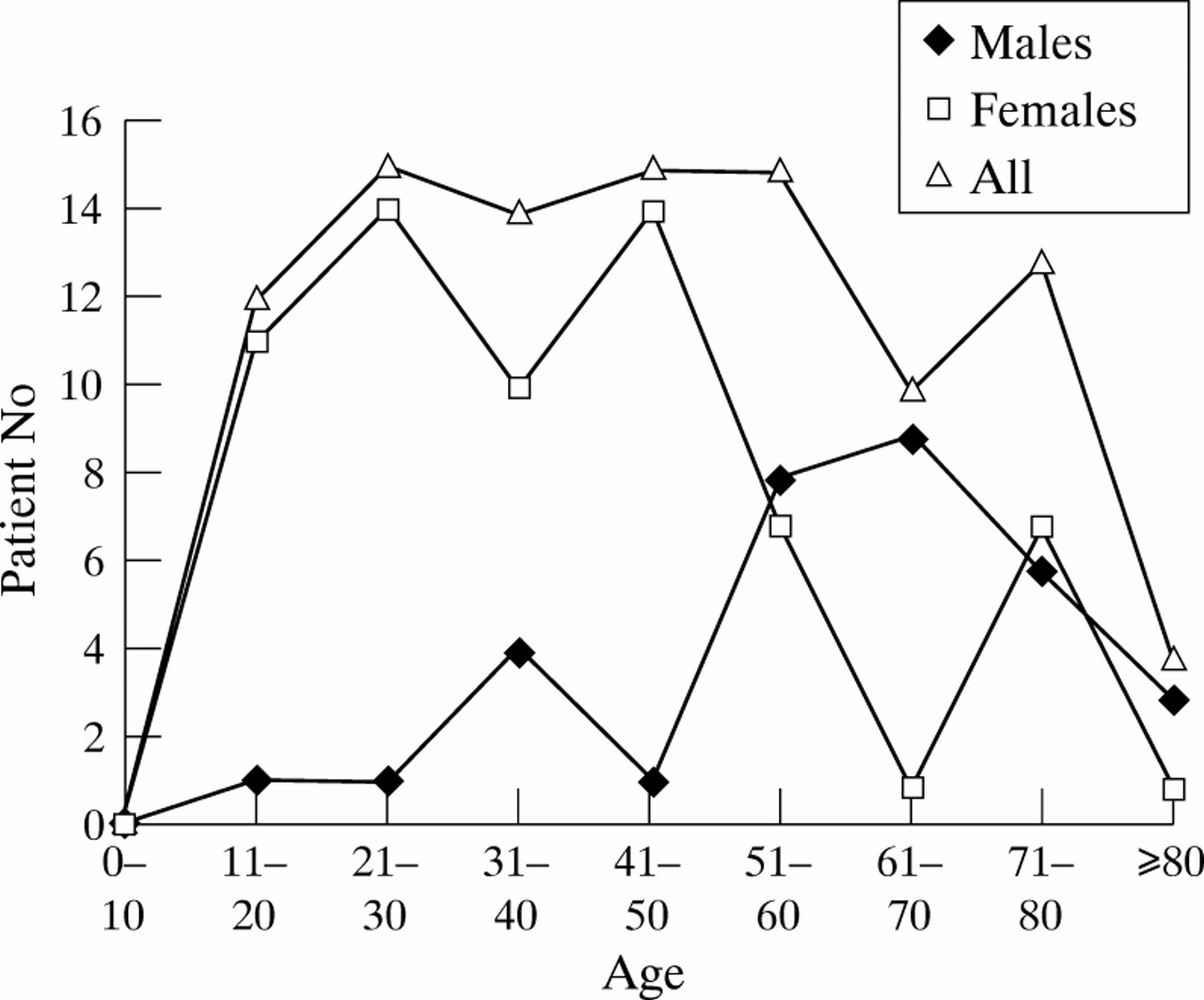
{kind=link}
Age specific onset for 100 prevalent patients.
Age and sex specific prevalence
INCIDENCE
In a five year period between 30 June 1992 and 1 July 1997 Cambridge based neurologists made 58 new diagnoses of myasthenia gravis, of which 38 lived within the study area; this provides a crude incidence of 11.1/million population/year.
CLINICAL CHARACTERISTICS
At presentation, 52/100 patients had ocular and 48/100 more generalised disease. The commonest presenting symptoms and signs were ptosis (64%) and diplopia (64%). In six (6%) patients, the delay from presentation to diagnosis was >1 year and initial erroneous diagnoses included motor neuron disease (n=1), dysthyroid eye disease (n=1), functional (n=2), or vocal dystonia (n=1). One patient was lost to follow up. Eight/100 (8%) patients had precipitating factors for initial disease expression of which the commonest were drugs (n=4), pregnancy (n=2), or infection (n=2). Eighty two/100 patients were known to have undergone a tensilon test at presentation and in 76/82 (93%) there was an unequivocal response. In 62/100 patients the results of acetylcholine receptor antibody assays were documented and positive in 50/62 (79%; male 18/23; 78: female 32/39; 82%). The mean disease duration was 13.7 years, and mean duration of follow up 11.9 years. Forty two/100 (42%) patients were under current neurological review and four were under the care of other specialists (ophthalmology (n=2) and general medicine (n=2)).
COURSE
Seventy five/100 patients had developed generalised disease at some stage and in 21/75 (28%) this was restricted to oculobulbar or facial symptoms. Of the fifty two patients who presented with pure ocular disease, 27/52 (52%) had gone on to develop generalised myasthenia. Patients had been admitted on average 1.9 times for investigation or control of disease. Fifty eight/100 had received immunosupressive therapy for a mean of 4.9 years. Fifty seven had received oral steroids, 17 azathiaprine, and only three plasmapheresis. Clinical classification according to the Osserman criteria identified 26/100 patients with grade I, 17 with grade IIa, 30 with grade IIb, II with grade III, and 16 with grade IV disease (table 3). The commonest side effects of treatment were anticholinergic effects of anticholinesterases severe enough to cause treatment modification (n=12), cataracts (n=3), recurrent infections (n=2), azathiaprine related neutropoenia (n=2), and osteoporosis (n=4). Twenty seven/100 (27%) patients had an additional history of autoimmune disease (males 2/33, 6%; females 25/67, 38%, table 4).
Diagnostic classification in 100 prevalent patients with myasthenia gravis
Cosegregation of autoimmune disease in 100 prevalent patients with myasthenia gravis
At the time of last review, 45/100 patients were asymptomatic. Sixty four/100 were currently taking anticholinesterases and 41/100 were being treated with immunosupressive therapy. Thirty six/100 thought that their disease had represented a major handicap interfering with social or professional life.
Thirty five/100 patients (males 8/35, 23%; females 27, 77%) underwent a thymectomy after a mean delay of 0.8 years from presentation. Patients with thymoma related myasthenia tended to have more severe disease than non-thymomatous patients (Osserman grade III or IV 7/13; 53%, table 5). Mean age at thymectomy was 27.8 years for those without and 39.6 years for those with a thymoma. Twelve patients had a thymoma and in three myasthenic symptoms postdated thymectomy by a mean of 2.2 years. One of 12 (8%) patients who had undergone thymectomy for thymoma and 6/22 (27%) without thymomas were currently in remission (stable and asymptomatic off treatment). This compares with 3/41 (7%) patients presenting with generalised disease who did not undergo thymectomy and 6/25 (24%) presenting with ocular disease (table5).
Thymectomy in 100 prevalent patients with myasthenia gravis
Discussion
It is generally held that the frequency of myasthenia is low irrespective of geographical, cultural, or racial boundaries. However, epidemiological surveys show considerable variation from 1.2/100 000 in Japan in 19584 to 17.5/100 000 in Cyprus in 19945; even contemporary studies performed over the past 10 years in developed countries show a threefold difference in prevalence which is unlikely to be explained solely by study methodology and suggests a trend towards rising prevalence over time (table 6).1
Comparison of contemporary population based studies
Here we demonstrate one of the highest prevalence and incidence figures for myasthenia, although our results are in line with recent large population studies from developed countries elsewhere in the world. Confirmation that our estimate of 15/100 000 population represents an increase in disease frequency compared with previous studies from the United Kingdom is made difficult because detailed earlier epidemiological information is lacking. The probable increase is likely to result from increased survival, an increasingly aged population who are at relatively higher risk of developing the disease, a genuine rise in incidence, the application of improved diagnostic tests, and increased recognition of milder cases in which ocular symptoms predominate—features which are all known to inflate prevalence in other neurological illnesses.6
Previous reports of myasthenia in the United Kingdom are almost entirely based on the personal experience of the individual neurologist with a particular interest in the disease, or on patients with a selected feature such as thymectomy, each of which is unlikely to be representative. The first epidemiological report from the United Kingdom was from Ferguson et al 7 in 1955, who reported on 85 patients diagnosed between 1932 and 1954 from a population of 4.5 million. Later an analysis of 60 patients from the Leeds area8 diagnosed between 1934 and 1955 determined a prevalence of 26/million and an incidence of five/million/year. A study of disease frequency in the Merseyside conurbation in 19619 found similar figures with a prevalence of 23/million and an incidence of 2.2/million/year. Finally Schon et al 10 in 1996 identified 22 patients newly diagnosed in Croydon over a 7 year period and, as in this study, found a large proportion (13/60, 22%) with onset over the age of 60, in marked contrast to Garland and Clark8 who identified only 3/60 (5%) in this age group.
In all these studies the detailed information required to analyse the impact of improved survival and a rise in incidence or prevalence over time is lacking. In addition, modern epidemiological methodology has undoubtedly improved case recognition. For all these reasons, direct comparison with earlier studies is problematic.
Elsewhere, three large studies are notable for their ability to provide longitudinal incidence data. In a series of detailed studies of the epidemiology of myasthenia in Norway from 1950 to 198211 a rise in prevalence was noted from 2.1 to 9.0/100 000 but a relatively stable incidence ranging from 2.1 to 4.1/million/year suggesting that the frequency of diagnosing new cases had not changed during the period of the survey. A similar pattern was noted in Denmark,12with prevalence rising from around 20/million in 1970 to 77/million in 1988 whereas incidence was stable at 4.4/million/year. Lastly, a 25 year survey of myasthenia in Sardinia13 saw prevalence rise from 7.7 to 45.0/ million between 1961 and 1986 with incidence at 2.5/million/year over the same period. Our study suggests a different interpretation. The error is usually to produce a minimum figure when incidence data is derived retrospectively from year of diagnosis and over a relatively short period. Clearly the change in incidence in the United Kingdom from 5 and 2.2 in the mid-1950s to 11.1/million in this study seems to indicate that incidence is also contributing to the rise in disease frequency. One striking difference in the subclassification of disease seen in most contemporary studies including our own is the high frequency of ocular disease; this accounted for only 12% in earlier Scandinavian surveys12 and 14% in a recent analysis of 100 consecutive patients from a neuromuscular clinic in the Netherlands.14 This difference may account for the higher overall prevalence in Cambridgeshire and in Virginia, North America.15
In summary, we have shown a high prevalence of myasthenia gravis in a previously unsurveyed part of the United Kingdom with nearly a third of cases having onset after the age of 60 years. Disease frequency seems to be rising over time and whereas this is likely to result from improved survival and the advent of new diagnostic techniques, it depends mainly on rising incidence in an increasingly aged population.