Article Text

Abstract
OBJECTIVE To determine the neuroradiological abnormalities associated with subjects carrying the mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS) tRNALeu(UUR)A3243G point mutation
METHODS Mitochondrial genetic analysis was performed on 24 subjects from six kindreds with the MELAS tRNALeu(UUR) A3243G point mutation. Cerebral CT and MRI were performed on 24 patients and 15 patients respectively. Previous neuroradiological investigations including cerebral CT from four deceased members of the families were also reviewed. Histological examination of postmortem specimens of two patients within the kindreds was performed.
RESULTS The commonest radiological finding was basal ganglia calcification. Other abnormalities included focal lesions and cerebellar and cerebral atrophy. Basal ganglia calcification was progressive, symmetric, and asymptomatic. Histologically, basal ganglia calcification in one patient was found to be in the pericapillary regions of the globus pallidus, with no neuronal involvement. Focal lesions most commonly involved the grey matter of the parietal and occipital lobes and cerebellum. Histopathological examination suggested that these were due to cellular rather than vascular dysfunction. Enlargement of the fourth ventricle was the first sign of cerebellar atrophy. Cerebral and cerebellar atrophy were only present with severe disease.
CONCLUSIONS These radiological findings, when considered in the context of the clinical and pathological findings, seem to reflect two major disease processes: an intermittent abrupt loss of function associated with cell injury from which there is at least partial recovery and a slowly progressive degenerative process causing basal ganglia calcification, and cerebral and cerebellar atrophy. The clinical and radiological features resulting from these processes are distinctive and provide insight into the consequences of mitochondrial dysfunction on the brain.
- MELAS
- A3243G point mutation
- mitochondrial DNA
- neuroradiology
Statistics from Altmetric.com
The mitochondrial cytopathies are a group of disorders defined by biochemical, morphological, and genetic abnormalities that are difficult to classify on clinical grounds alone.1 Three main clinical syndromes in adults are recognised: mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS), myoclonic epilepsy and ragged red fibre disease (MERRF), and Kearns-Sayre syndrome (KSS).2 The overlap in the clinical expression of these syndromes was the subject of a previous report.3 It is now possible to classify some of these disorders on a genetic basis: MELAS is associated with point mutations involving, among others, the leucine transfer RNA at nt position 3243,4 3271,5 and ND4 subunit at nt 110846; MERRF at nt position 83447 and 83568; and KSS with large scale deletions of up to 9kb in length most commonly involving regions between the ATPase 8 and ND5 genes.9
The neuroradiological abnormalities associated with the mitochondrial cytopathies such as intracerebral calcification, focal hypodensities, and generalised atrophy are characteristic and are useful in the clinical diagnosis of these disorders. Previous reviews of these abnormalities have included only a few patients or a diverse array of mitochondrial disorders,10-13 leaving the relation to specific mitochondrial genetic abnormalities unclear. The aim of this study was to determine the neuroradiological features of many patients with the same genetic abnormality and to relate these to the patients’ symptoms, signs, and pathology.
Patients and methods
PATIENTS
After the identification of six probands who presented with MELAS syndrome, 24 living family members, at risk through a maternal line of inheritance, agreed to participate in the study. All members were examined by a neurologist and their clinical features documented. Four deceased family members who had documented clinical features suggestive of MELAS syndrome and in whom radiological information was also available were included in the study. The project was approved by the Western Sydney Area human ethics committee and written consent was obtained from all subjects or if deceased, their next of kin.
METHODS
Mitochondrial genetic analysis
Total DNA from muscle biopsy, blood, and/or hair follicle samples from each patient was digested by proteinase K at a final concentration of 200 μg/ml in a buffer solution containing 50 mM Tris HCl pH 8.5, 1mM EDTA, and 0.5% Tween 20 overnight at 37°C. MtDNA was amplified using oligospecific primers as described previously.14 The polymerase chain reaction (PCR) product (902bp in length) was digested with Apa I overnight, run on a 1% agarose gel, and photographed under UV light. As the mutant mtDNA creates an additional restriction site for Apa I, those patients with the MELAS A3243G point mutation had two additional bands (416 and 486 bp) identified.
Neuroimaging
Cerebral CT with 5–10 mm slices was performed on all patients who were available for study and 28 age and sex matched controls. Controls were from a series of unselected patients who had cerebral CT for various reasons at Wesmead Hospital in January 1996 and in whom no intracranial pathology was found. Axial slices 5 mm thick were obtained in the region of the posterior fossa with the scans angled parallel to the radiographic baseline: a line drawn from the outer canthus of the eye to the external auditory meatus. Intravenous contrast was given if there was evidence of a focal lesion. Mineralisation of the globus pallidus, putamen, caudate, thalamus, and dentate nuclei was categorised as “basal ganglia” calcification. To assess enlargement of the fourth ventricle, the maximal width of this structure was measured on axial CT at the level of the pons. Generalised cerebellar atrophy was defined as enlargement of the superior vermian cistern, the cerebellopontine angle, and fourth ventricle and prominence of the cerebellar folia. Cerebral atrophy was defined using Evans’ ratio15: the greatest width of the frontal horns of the lateral ventricles divided by the width of the internal diameter of the skull at the level >0.30–0.33 (depending on the age of the patient). All previous available cerebral CT was reviewed. In nine patients, CT had been repeated over periods ranging from 4 to 7 years, and one patient had a series of CT spanning 10 years (table 1). Fifteen patients had cerebral MRI. T1 and T2 weighted images were obtained and images were reconstructed in the sagittal and axial planes.
Clinical symptoms and signs of the patients studied
Histopathology
Postmortem specimens were examined from two patients from two kindreds (1.IV.4 and 3.II.1). Brains were fixed in formalin and 7 μm paraffin sections from multiple brain regions were stained with haematoxylin and eosin.
Results
Figure 1 shows the relevant sections of the family pedigrees and table 1 shows their clinical symptoms and signs.

Family pedigrees of the six families identified with the MELAS A3243G point mutation. Patient identification numbers are the same as those that appear in the text.
MITOCHONDRIAL GENETIC ANALYSIS
Twenty two patients had the MELAS point mutation A3243G identified on hair, blood, and/or muscle. Four patients had the clinical features of MELAS syndrome but were not tested (before their death) for the MELAS A3243G point mutation. Two other patients did not have the genetic abnormality identified and were subsequently excluded from the study.
RADIOLOGICAL FEATURES
The radiological features found in the 26 patients included basal ganglia calcification, enlargement of the fourth ventricle, focal lesions, cerebral atrophy, and cerebellar atrophy (table 2).
Neuroradiological findings on CT of the 26 patients studied
Basal ganglia calcification
Basal ganglia calcification was found in 14 patients; this affected the globus pallidus in 12 patients. Calcification was also seen in the putamen, caudate, and thalamus (fig 2). In two patients, there was also calcification in the corona radiata. Prominent calcification was seen in both young and old patients (age range 17–76 years, mean age 54 years), with nine out of the 14 patients being under 60. In four patients, calcification was absent on the first scan but present on a repeat scan 4–5 years later. No patient had clinical features to suggest basal ganglia dysfunction. The serum calcium concentration in all 14 patients was normal. The serum parathyroid hormone concentration, in six patients in whom it was measured, was also normal.

CT showing basal ganglia calcification: (A) in the globus pallidus, putamen, and caudate of a 24 year old woman (4.II.1), (B) thalamus of a 48 year old man (3.II.1).
Enlargement of the fourth ventricle and cerebellar atrophy
The mean diameter of the fourth ventricle in the age and sex matched controls was 10.9 (SD 1.2, range 8–14) mm. Defining enlargement of the fourth ventricle as >15 mm (mean +3 SD), isolated enlargement of the fourth ventricle was found in nine patients (fig 3A). Patients with enlarged fourth ventricles were aged between 12 and 68 years; eight of the nine were under 60 years old (tables 1 and 2). Two patients with isolated enlargement of the fourth ventricle on initial scanning showed generalised cerebellar atrophy when the scans were repeated 4 years later. Four patients had generalised cerebellar atrophy; all had a wide based gait and mild limb ataxia. None of these patients had a history of alcohol misuse and only one patient had taken phenytoin or other drugs that may have caused cerebellar atrophy.

Evolution of a CT series of patient 1.IV.3 spanning a period of eight years showing (A) bilateral basal ganglia calcification and enlargement of the fourth ventricle at the age of 17 years, (B) four months later when comatose, showing bilateral cerebellar swelling and hypodensity causing secondary obstructive hydrocephalus due to obliteration of the previously enlarged fourth ventricle, (C) left parieto-occipital hypodensity associated with right hemiparesis, right hemianopia, and aphasia at the age of 21 years, (D) right sided parietal lesion associated with a left hemiparesis at the age of 25 years. In the later scans, the patient’s basal ganglia calcification becomes more prominent and cerebral atrophy develops.
Focal lesions
In nine patients, discrete hypodense lesions were seen on CT. In eight of these, the lesions involved the parietal and occipital lobes. One patient also had hypodense lesions of both cerebellar hemispheres. Figure 3 shows a series of scans from a patient with multiple stroke-like episodes. In four patients, the lesions were not confined to the territory of a single major artery, such as the middle or posterior cerebral arteries or their branches. Cerebral angiograms performed in four patients were normal with no abnormality in the arterial or venous phase of the studies.
The lesions seen on CT usually involved the grey and white matter. In the acute stage, there was enhancement with intravenous contrast and in some cases, a mass effect. Cerebral MRIs showed increased signal intensity on T2 weighted images, mainly within the grey matter of the cortical mantle.
All patients with focal lesions on CT had a history of stroke-like episodes (table 3). These were characterised by a clinical prodrome followed by a sudden neurological deficit. The prodrome consisted of subacute onset of nausea, vomiting, throbbing headache, and abdominal pseudo-obstruction (in varying combinations) and would commence hours to days before the neurological deficit. The prodrome was usually the same for each patient but varied from patient to patient. A neurological deficit, such as hemiplegia, hemianopia, ataxia, or aphasia would follow. There were 12 episodes in the nine patients who had focal lesions; 10 of these events were preceded by a clinical prodrome. Six of the nine patients had a history of at least one prodrome which did not proceed to a neurological deficit. In five patients, these stroke-like episodes were associated with seizures, both focal and generalised. In each case, the site of the focal lesion seen on neuroimaging was appropriate for the neurological deficit. In five instances, stroke-like episodes followed a metabolic or toxic stress; these included an infected toe nail, heat stroke, gastroenteritis, or viral illness.
Clinical features of patients who had focal lesions seen on CT
Seven of the nine patients required admission to hospital during the stroke-like episode. There was usually good clinical recovery within six months. One patient (5.II.1), with an episode of dense hemiparesis and hemianopia associated with the appearance of a hypodense region involving the right parieto-occipital area on CT, made a full recovery within a few weeks; MRI a month after the event showed only minor changes. Seven of the nine patients with focal lesions also had basal ganglia calcification; the two without were both aged younger than 20. Three patients who had stroke-like episodes eventually developed cerebral atrophy and cognitive impairment.
Cerebral atrophy
Cerebral atrophy was present in six patients, four of whom were younger than 40 at the time of scanning (range 17–76 years). The Evans’ ratio was highest in those patients with multiple episodes of neurological deficits. Five of the six patients with cerebral atrophy had basal ganglia calcification. All had an onset of symptoms before the age of 40.
HISTOPATHOLOGY
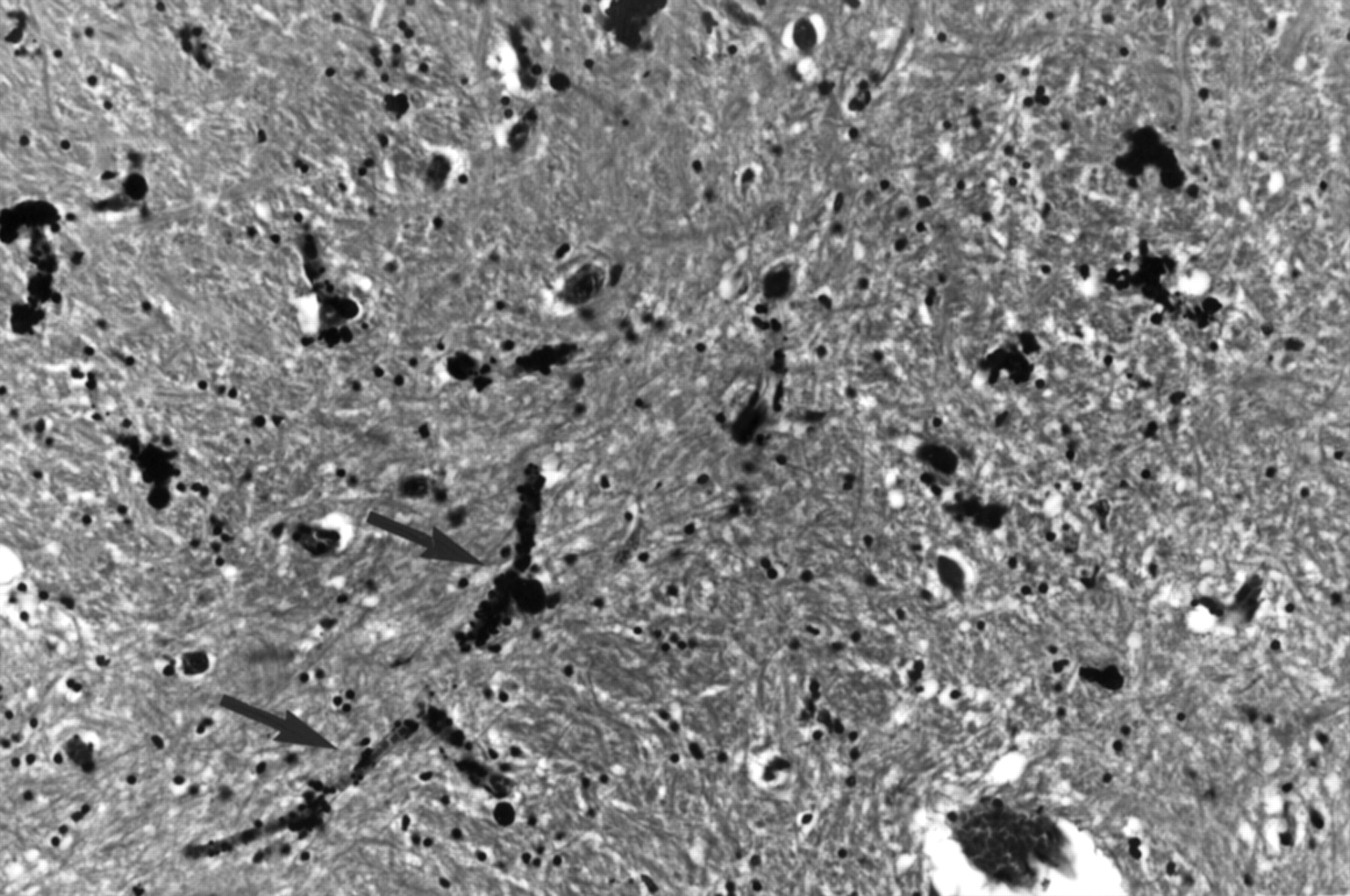
Postmortem specimens were available in two patients. In patient 1.IV.4, there was extracellular calcification in the pericapillary regions of the globus pallidus. There were tubular casts of mineral deposition within the blood vessel walls. No calcification was seen within neurons (fig 4).
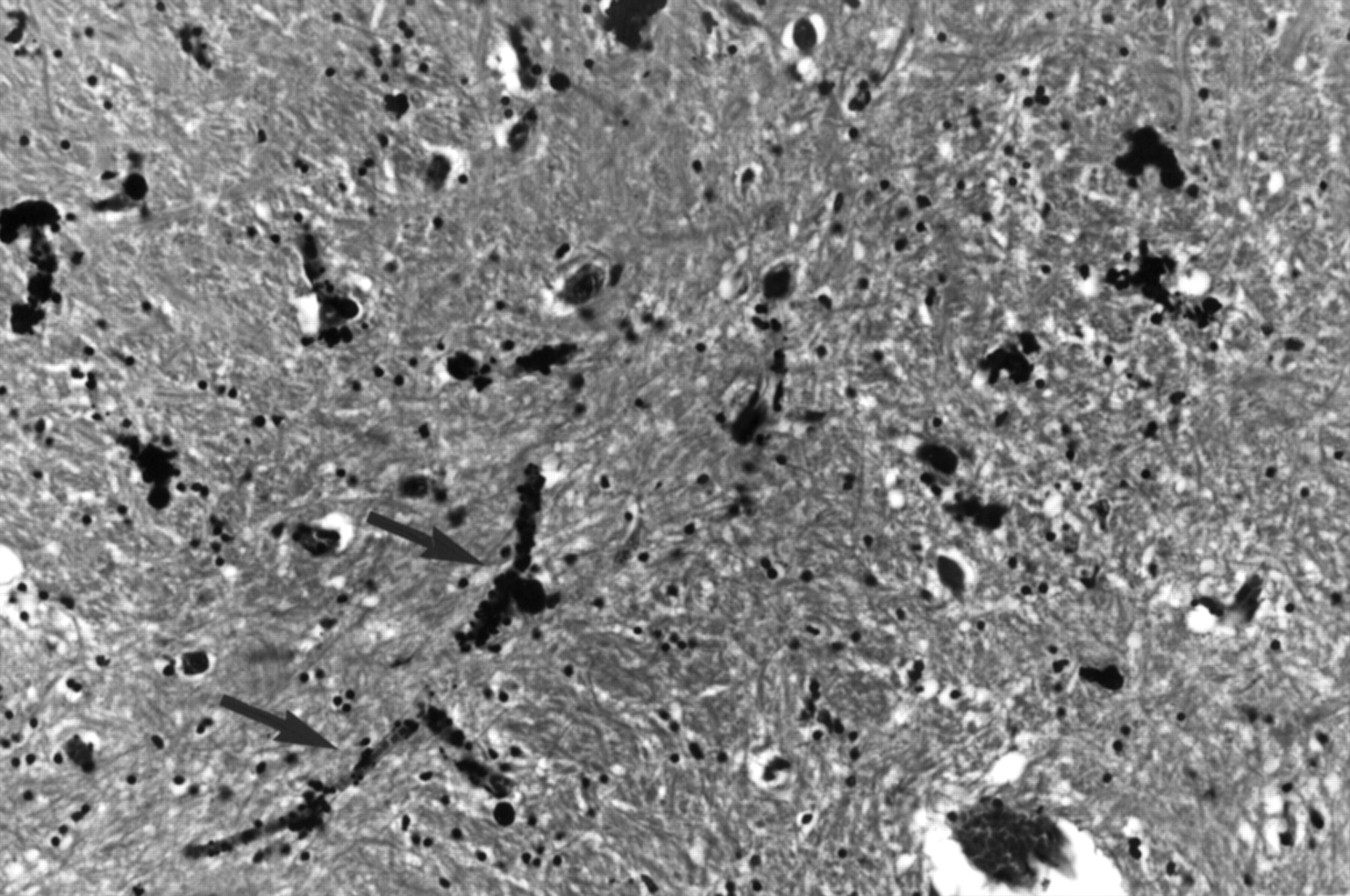
Haematoxylin and eosin section of the globus pallidus of patient 1.IV.4 showing tubular casts of mineral deposition within the capillary walls (arrows). No neuronal calcification is seen. (Magnification×45.)
Focal lesions disclosed areas of cell loss in a laminar distribution of cerebral cortex. This predominantly involved layers 3 and 5 of the cortex. Within these lesions there was spongiform change, astrocytic proliferation, capillary proliferation and relative neuronal sparing (fig 5). The white matter was affected minimally. In one patient (3.II.1) the hippocampus was histologically normal, in the other patient (1.IV.4) there was cell loss in the endfolium of the hippocampus only.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Magnified view (×10) of the parietal cortex of patient 1.IV.4 showing laminar distribution of necrosis, predominantly involving layers 3 and 5 (arrow) and (B) magnified view (×200) of a cortical lesion in the insular region of patient 1.IV.4 showing spongiform change, astrocytic proliferation, capillary proliferation, and relative neuronal sparing. A preserved neuron is indicated by the arrow.
Discussion
Neuroradiological abnormalities in this study included calcification of the basal ganglia, enlargement of the fourth ventricle, and focal lesions, which usually involved the parieto-occipital regions of the cerebral hemispheres. Late features included cerebral and cerebellar atrophy. The neuroradiological picture was distinctive and highly suggestive of MELAS syndrome when all these features were present. The discussion deals with each feature in turn.
BASAL GANGLIA CALCIFICATION
In the present study, basal ganglia calcification was the commonest (54%) neuroradiological abnormality in patients with the MELAS A3243G point mutation. This compares with an incidence of 13% in a series of 820 unselected patients who had cerebral CT for various indications performed at our hospital (unpublished data). Basal ganglia calcification in our patients was progressive, symmetric, and most commonly involved the globus pallidus. It also affected other areas such as the caudate, putamen, thalamus, and internal capsule, but unlike previous studies,13 was not seen in the dentate nucleus. Hypoparathyroidism, an important cause of basal ganglia calcification16 and one which may occur in association with mitochondrial disorders,17 was not present in any of the patients in this study.
Basal ganglia calcification in our patients was in the same histological18 and radiological distribution as in normal aging brains, but often occurred at an earlier age. As mitochondrial dysfunction occurs with aging,19 it is tempting to speculate that deposition of minerals in the basal ganglia in the normal population is due to cumulative mitochondrial defects associated with aging. The fact that basal ganglia calcification occurs after a wide range of insults to the brain such as hypoxia,20radiation,21 and congenital hypothyroidism,22suggests that this part of the brain has a propensity to respond to injury or metabolic disturbance by laying down minerals.23Why basal ganglia calcification occurs in patients with mitochondrial disease is unclear. Mitochondria are important in the regulation of intracellular calcium concentration24 and it has been suggested that calcium may accumulate within the mitochondria and deposit as salts during periods of stress.25 Defects in the mitochondrial genome may impair the mitochondrion’s ability to function, resulting in calcium deposition in susceptible regions.
None of the patients with basal ganglia calcification showed signs of basal ganglia dysfunction such as Parkinson’s disease or dystonia. In the one patient with basal ganglia calcification studied at postmortem, no mineral deposition within the neurons of the basal ganglia or neuronal loss within these regions was found although neuronal loss associated with basal ganglia calcification has previously been reported.26 Although extrapyramidal symptoms have occasionally been described in patients with basal ganglia calcification27 28 most patients are asymptomatic.29 30 Normal basal ganglia function may be explained by this neuronal sparing, as seen in our patients. In addition, the lack of symptoms may relate to the functional reserve of the extrapyramidal system resulting from its organisation as a parallel processing system.31 Basal ganglia calcification is thus a useful marker of mitochondrial disease though it does not usually cause clinical symptoms.
FOURTH VENTRICLE ENLARGEMENT AND CEREBELLAR ATROPHY
Enlargement of the fourth ventricle was found more often than generalised cerebellar atrophy. Isolated enlargement of the fourth ventricle was often seen in young patients, some of whom were asymptomatic, but generalised cerebellar atrophy was only seen in those patients with longstanding (>10 years) disease. Isolated enlargement of the fourth ventricle developed into generalised cerebellar atrophy in two patients. Thus this feature seems to be the first sign of cerebellar atrophy in patients with MELAS. Selective atrophy of the anterior superior vermis, the structure which forms the roof of the fourth ventricle, has been reported in pyruvate dehydrogenase deficiency, a disorder which produces a similar metabolic derangement to the mitochondrial cytopathies.32 The anterior superior vermis seems to be the first part of the cerebellum to atrophy in MELAS and the resultant fourth ventricle enlargement may thus be regarded as an early, though not specific, marker of disease.
CEREBRAL ATROPHY
All patients with cerebral atrophy had longstanding disease. Only half of the patients had a history of multiple stroke-like episodes. It seems likely that atrophy results from both sudden loss as well as gradual attrition of cells.33 34 The progressive atrophy may result from increased release of free radicals from the respiratory chain in genetically damaged mitochondria.35
FOCAL LESIONS
Focal lesions found on CT usually involved the parietal and occipital lobes. This distribution has been previously noted2 36 37 but why this region is preferentially affected remains unclear. Cells in this area might be expected to carry a higher proportion of mutant to wild type mitochondrial DNA but quantitative studies, performed on postmortem specimens,38-40 have failed to confirm this. The preferential involvement of the parieto-occipital region highlights the similarities between mitochondrial disease and migraine: both also have clinical prodromes of nausea, vomiting, and visual scintillations and are associated with headache. In migraine, a wave of focal hypoperfusion progressing from the occipital to parietal regions41 42 occurs during the aura, causing the typical visual symptoms. The hyperaemia which follows the migraine aura has also been described in patients with MELAS.43-44
Focal lesions were always associated with a history of stroke-like episodes. Although the sudden onset of these episodes is suggestive of a vascular mechanism,45 the lesions often crossed adjacent arterial territories (sometimes involving posterior and anterior circulations) making it unlikely that these lesions resulted from large vessel occlusion. As the stroke-like episodes were often preceded by toxic or metabolic stressors that may have precipitated dehydration, the possibility of venous thrombosis could also be raised. However, the appearance and distribution of the focal lesions seen in the present study differed from those of venous infarcts which are often haemorrhagic and usually multifocal or bilateral.46Carotid angiograms performed in this study and others26 33 47 failed to show either arterial or venous abnormalities.
Involvement of the arterioles in the pathogenesis of the stroke-like episodes has been postulated.45 48-50Goto45 speculated that arterioles, strongly reactive to succinic dehydrogenase,50 51 contract abnormally in patients with MELAS. This mechanism would lead to a reduction in blood supply, thus leading to a stroke-like episode. This theory is yet to be substantiated, as occlusion of cerebral arterioles has not been shown in MELAS. Furthermore, arteriolar disease usually results in small subcortical white matter lesions52 rather than the selective cell loss in the grey matter seen in our patients.
Histopathological examination of the focal lesions disclosed a laminar distribution of neuronal cell loss, changes often seen with global hypoperfusion, such as occurs during a cardiac arrest. The changes, however, differed from global hypoperfusion in several ways: the lesions were scattered throughout the cortical rim and were of differing ages; there was neuronal sparing amongst the areas of astrocytic proliferation and spongiform change instead of confluent areas of necrosis; and the hippocampus, which is almost always affected by global hypoperfusion, was entirely normal in one patient and atypically affected in the other. There was no history of cardiac arrest or prolonged hypotension in either patient. These findings suggest that the focal lesions do not result from episodes of global hypoperfusion.
Focal lesions predominantly affected the grey matter, suggesting that the lesions may result from a sudden inability of cortical cells with abnormal mitochondria to cope with the metabolic demands placed on them. In support of this argument recent studies using MR spectroscopy have shown increased concentrations of lactate11 and impaired oxidative metabolism within the focal cortical lesions during an acute episode, with return to normal after clinical resolution.34 PET studies have shown a reduction in the oxygen extraction rate in the presence of preserved cerebral blood flow during an acute event, particularly in the grey matter of the posterior cortex.53 Preserved or increased cerebral blood flow has also been found on both SPECT43 44 and xenon enhanced CT,47 suggesting that there is a compensatory vascular response to a metabolic stress. Hence, the vascular changes seem to be secondary to the metabolic disturbance taking place in the parenchyma of the grey matter.
Perhaps the sudden imposition of a biological stress, such as infection, leads to a failure of mitochondrial function in susceptible neurons in the grey matter. A cascade of biological events then occurs with accumulation of cellular metabolites which causes regional hyperaemia. The combination of regional hyperaemia and toxic cellular metabolites may be responsible for the hyperexcitability leading to visual scintillations, seizures, nausea, and vomiting found in the prodromal phase. As the metabolic derangement becomes more severe, cellular dysfunction predominates, resulting in a neurological deficit. Clinical improvement, which is such a feature of these episodes,37 54 55 occurs with recovery of mitochondrial function, with the ultimate deficit being dependent on the proportion of neurons surviving the metabolic insult.
Focal lesions in the parietal and occipital lobes in the presence of cerebral and cerebellar atrophy and basal ganglia calcification provide a distinctive neuroradiological picture which should raise the suspicion of a mitochondrial disorder, particularly in a young patient.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia and the Winthrop Fellowship. We also thank Dr W Evans for his help with obtaining pathological specimens used in this work.