Article Text

Abstract
OBJECTIVES AND METHODS Seven families were studied with an axonal form of Charcot-Marie-Tooth disease (CMT) associated with mutations in the peripheral myelin protein zero (MPZ) gene—Thr124Met or Asp75Val.
RESULTS Patients with these mutations commonly showed relatively late onset sensorimotor neuropathy predominantly involving the lower limbs. Sensory impairment typically was marked, and distal muscle atrophy and weakness were also present in the legs. Adie's pupil and deafness were often present, and serum creatine kinase concentrations were often raised irrespective of which MPZ mutation was present. Relatively well preserved motor and sensory nerve conduction velocities contrasted with reduced or absent compound muscle action potentials and sensory nerve action potentials. Axonal change with marked axonal sprouting was seen in sural nerve specimens.
CONCLUSION The similar associated clinical findings suggest that patients with axonal CMT with an MPZ gene mutation share distinctive clinical features.
- Charcot-Marie-Tooth disease type II
- myelin protein zero
- Adie's pupil
Statistics from Altmetric.com
Charcot-Marie-Tooth disease (CMT) type 2 is a familial axonal neuropathy currently known to include several genetic subtypes assigned to multiple gene loci,1-3 but the specific mutation causing the neuropathy has not been identified. Mutations of the peripheral myelin protein zero (MPZ) gene has been reported to cause some cases of CMT type 1B4 5 a representative demyelinative form of CMT. In 1998, Marrosu et al 6 reported an axonal phenotype of CMT associated with an MPZ mutation (Ser44Phe). In 1999, De Jonghe et al 7 and Chapon et al 8 reported families with another MPZ mutation (Thr124Met) that exhibited an axonal phenotype characterised by late onset and marked sensory abnormalities, and in some families also deafness and pupillary abnormalities. Subsequently, several Europian families with axonal phenotype and MPZ mutations including Ala76Val, Tyr119Cys, and Asp61Gly have been reported.9 10 These reports suggest that MPZ mutations can induce axonal as well as demyelinative phenotypes in CMT. However, a detailed associated clinical phenotype has not been well documented in these mutations other than Thr124Met. We describe seven families with an axonal form of CMT associated with two different MPZ mutations (Thr124Met or Asp75Val) that showed a similar distinctive late onset clinical phenotype including sensorimotor impairment, Adie's pupil, deafness, and increased creatine kinase in serum. Mutations of the MPZ gene that cause an axonal form of CMT may often be associated with a distinctive phenotype.
Patients and methods
Patients in this study were from seven Japanese families with an axonal form of CMT disease associated with a mutation in the MPZ gene. In four families the mutation was Thr124Met, and in three families it was Asp75Val. Axonal phenotypes were evident in the clinicopathological features shown by the probands in each of the seven families. Axonal features included relatively well preserved nerve conduction velocities (motor conduction velocities (MCVs)>38 m/s in the median nerve) by contrast with low amplitudes of compound muscle action potentials (CMAPs) or sensory nerve action potentials (SNAPs). When obtained, sural nerve specimens did not show “onion bulb” formation. Instead, abundant axonal sprouting was evident in sections and a low frequency of segmental demyelination was seen in teased fibre preparations. All patients underwent neurological examination, nerve conduction studies, a CSF examination, and routine blood and urine examinations. Three patients also underwent sural nerve biopsy. The nerve conduction study was performed by standard methods.11 Sural nerve specimens, fixed in 2% glutaraldehyde solution with 0.025% cacodylate buffer (pH 7.4), were processed for semithin and ultrathin sections and for teased fibre studies.12-14 Density of myelinated fibres was assessed directly from toluidine blue stained transverse semithin sections of sural nerve using a computer assisted image analyzer (Luzex FS, Nireco) as described previously.12-14Unmyelinated fibre density was assessed using the same system with randomly taken electron microscopic photographs (magnification×5000) of uranyl acetate stained transverse ultrathin sections.12-14 Isolated single nerve fibres obtained from specimens by teasing were evaluated pathologically according to criteria described previously.12 15 16
Analysis of DNA was performed after obtaining informed consent. Genomic DNA was extracted from leucocytes. We screened members of the seven families for mutations in the MPZ gene by a non-isotopic RNAase cleavage assay (NIRCA).17 We then performed direct DNA sequencing of the MPZ gene. Briefly, the MPZ gene was amplified as three fragments by polymerase chain reaction (PCR). Sequences of primers for the MPZ gene were as follows: forward primer for fragment 1 (F1), 5′-CTA GGG ATT TTA AGC AGG TTC C-3′ and reverse primer for fragment 1 (R1), 5′-ATT GCT GAG AGA CAC CTG AGT CC-3′ (for exon 1); F2, 5′-CCA TAG GTG CAT CTG ATT CC-3′; and R2, 5′-CCT CCT TAG CCC AAT TTA TC-3′ for (exon 2); and F3, 5′-CAG CTG TGT TCT CAT TAG GGT CCT C-3′ and R3, 5′-GCT CAT CCT TTC GTA GCT CCA TCT C-3′ (for exons 3 to 6). The T7 promoter sequence 5′-TAA TAC GAC TCA CTA TAG GG-3′ was attached to each primer at the 5′ end. Amplification by PCR was performed using AmpliTaq Gold (Perkin Elmer). Amplification conditions included an initial denaturation incubation at 95°C for 9 minutes, followed by 35 cycles of denaturing at 95°C for 1 minute, annealing at 60°C for 1 minute, and elongation at 72°C for 2 minutes. NIRCA was performed using a Mutation Screener kit (non-isotopic RNAase cleavage assay; Ambion) according to the manufacturer's protocol. RNase digested products were electrophoresed in 2.5% agarose gels. Polymerisation chain reaction products were purified from the unreacted primers and nucleotides using a QIAquick PCR purification kit (QIAgen) according to the manufacturer's protocol. The purified products were sequenced directly using a Thermo Sequenase Cy5.5 terminator kit (Amersham Pharmacia) and analyzed on a GeneRapid sequencer system (Amersham Pharmacia). Nla III (New England Biolabs) digestion for Thr124Met mutation and Hinc II (New England Biolabs) digestion for Asp75Val were performed on PCR amplified exon 3 and exon 2 respectively according to the manufacture's protocol. Digestion products were electrophoresed on 2% or 3% agarose gel stained with ethidium bromide.
Results
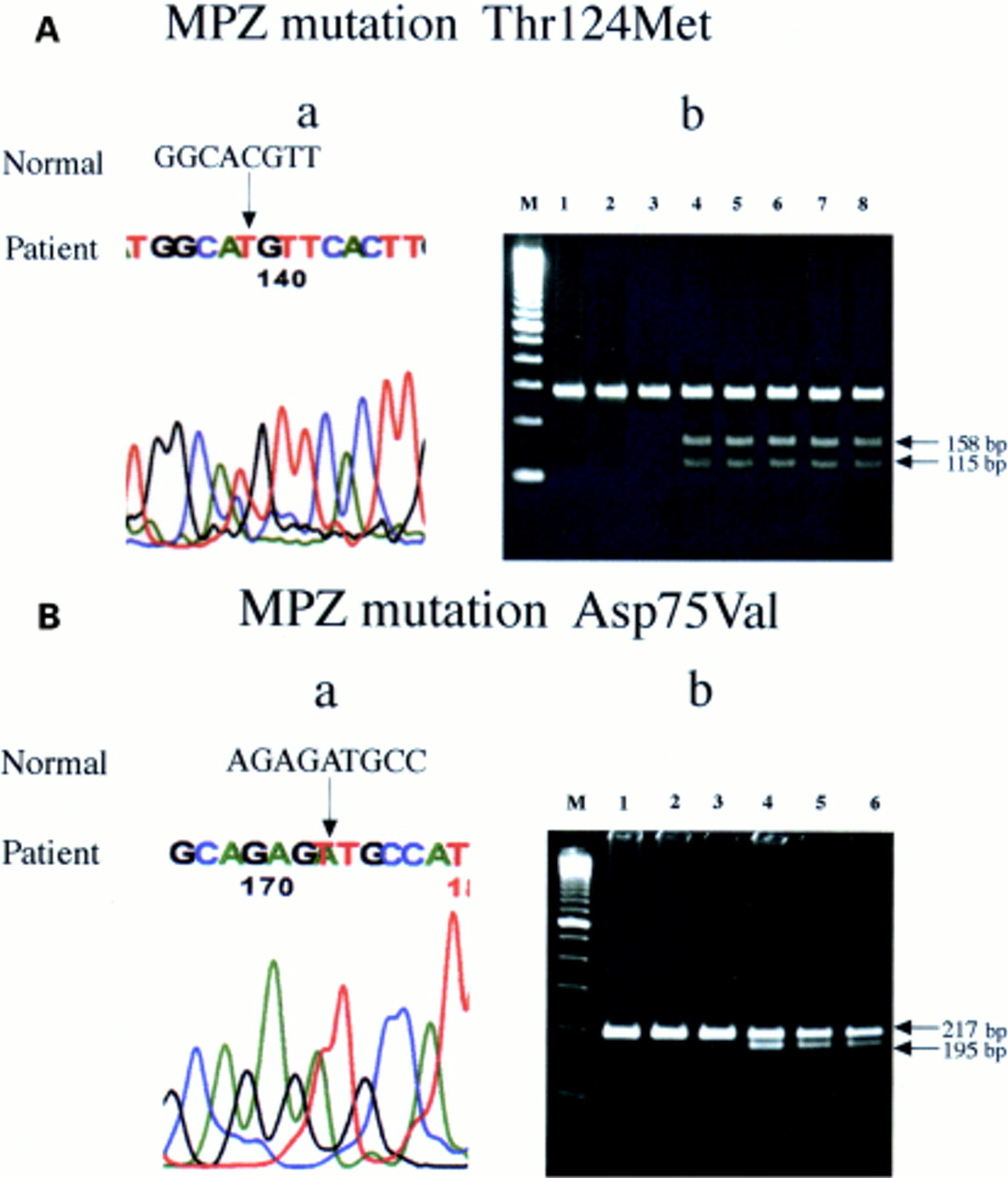
The PMP22 and C×32 genes were normal. One of two altered NIRCA patterns in the MPZ gene was detected in patients from the seven families, each resulting from a missense mutation. In four families a C to T substitution at codon 124 in exon 3 (ACG to ATG) resulted in an amino acid change from Thr to Met (Thr124Met) (fig 1 A a). The specific DNA fragment with this mutation was obtained by restriction digestion with Nla III (fig 1 A b). In three families an A to T substitution at codon 75 in exon 2 (GAT to GTT) was seen, resulting in an amino acid change from Asp to Val (Asp75Val) (fig 1 B a). The specific DNA fragment with this mutation was obtained by restriction digestion with Hinc II(fig 1 B b). The Asp75Val mutation represented a novel MPZ mutation.

(A a) Direct DNA sequencing of the MPZ gene. A C to T substitution at codon 124 in exon 3 (ACG to ATG) resulted in an amino acid change from Thr to Met (Thr124Met). (A b) Nla III digest of myelin protein zero gene exon 3. In normal controls (lane 1–3), no restriction fragment were obtained. In affected members (lanes 4–8) the 158 and 115 bp fragments were obtained (arrows). (B a) Direct DNA sequencing of the MPZ gene. An A to T substitution at codon 75 in exon 2 (GAT to GTT) resulted in an amino acid change from Asp to Val (Asp75Val). (B b) Hinc II digest of myelin protein zero gene exon 2 amplicons using the mismatch primer. In normal controls (lanes 1–3), no restriction fragments were obtained. In affected members (lanes 4–6), the 195 and 217bp fragments were obtained (arrows). The size marker M represents 100 bp.
ILLUSTRATIVE PROBANDS
Patient 1 (family 1)
In family 1 the proband case was a 47 year old man complaining of paraesthesias in the soles of both feet and leg cramps. He first noted cramps in the right calf muscles at age 37. Subsequently these became more frequent and prolonged, and bilateral plantar numbness developed. The patient had shown normal physical abilities for age in elementary and middle school.
Neurological examination showed bilateral pupillary dilation and extremely slow light and convergence reflexes, with miosis in response to 0.125% pilocarpine; this was diagnosed as Adie's pupil. Auditory acuity was slightly reduced for low tones. Paraesthesias were noted over both soles. Pain, light touch, and vibration perception were reduced in both legs below the knees, and joint position sense was slightly reduced. Muscle strength seemed normal in the upper limbs but was slightly reduced in both lower limbs. Amyotrophy of the left leg and hypertrophy of the right leg were seen. Toe walking was slightly impaired. Deep tendon reflexes were diffusely diminished. No pathological reflexes were found. A postural tremor involved the right hand.
In serum the creatine kinase concentration was slightly increased (362 IU/I). In CSF the protein concentration was raised to 87 mg/dl. For the median nerve the MCV was 44 m/s, and the amplitude of the CMAP was 11 mV. The tibial nerve showed an MCV of 35 m/s and a CMAP of 1 mV. The sural nerve exhibited a sensory conduction velocity (SCV) of 53 m/s and a SNAP of 4 μV. Amplitudes for CMAP and SNAP were extremely reduced in the legs, but conduction velocity was well preserved. Electromyography showed abnormalities suggesting chronic denervation. A sural nerve biopsy specimen (fig 2) disclosed moderate oedema of the endoneural and subperineural space and a moderate myelinated fibre loss that was most pronounced in large fibres. Marked axonal sprouting and atrophy were seen in residual myelinated fibres, and onion bulb formation was absent. Unmyelinated fibres were relatively preserved but showed sprouting. In assessment of teased fibres, 83% were normal, 10% showed axonal changes, and 7% showed remyelination. A biopsy specimen from the gastrocnemius muscle showed small angulated fibres, small grouped atrophy, and hypertrophic fibres, all representing chronic neurogenic changes. DNA analysis detected a C to T substitution at codon 124 in exon 3 of the MPZ gene, resulting in an amino acid change from Thr to Met (Thr124Met; fig 1 A a). The patient's older brother (II-1, fig 3 A family 1) noted paraesthesias in both legs at the age of 56. Subsequently, weakness, amyotrophy, and sensory impairment developed in the limbs. Adie's pupil was not present. An older sister (II-5, fig 3 A family 1) exhibited a right sided Adie's pupil, decreased muscle strength in the legs, and sensory impairment distal to both knees. Another older sister (II-7, fig 3 A family1) developed gait disturbance and apparent loss of muscle strength in the legs when aged about 50, with amyotrophy and sensory impairment and associated pupillary abnormality.
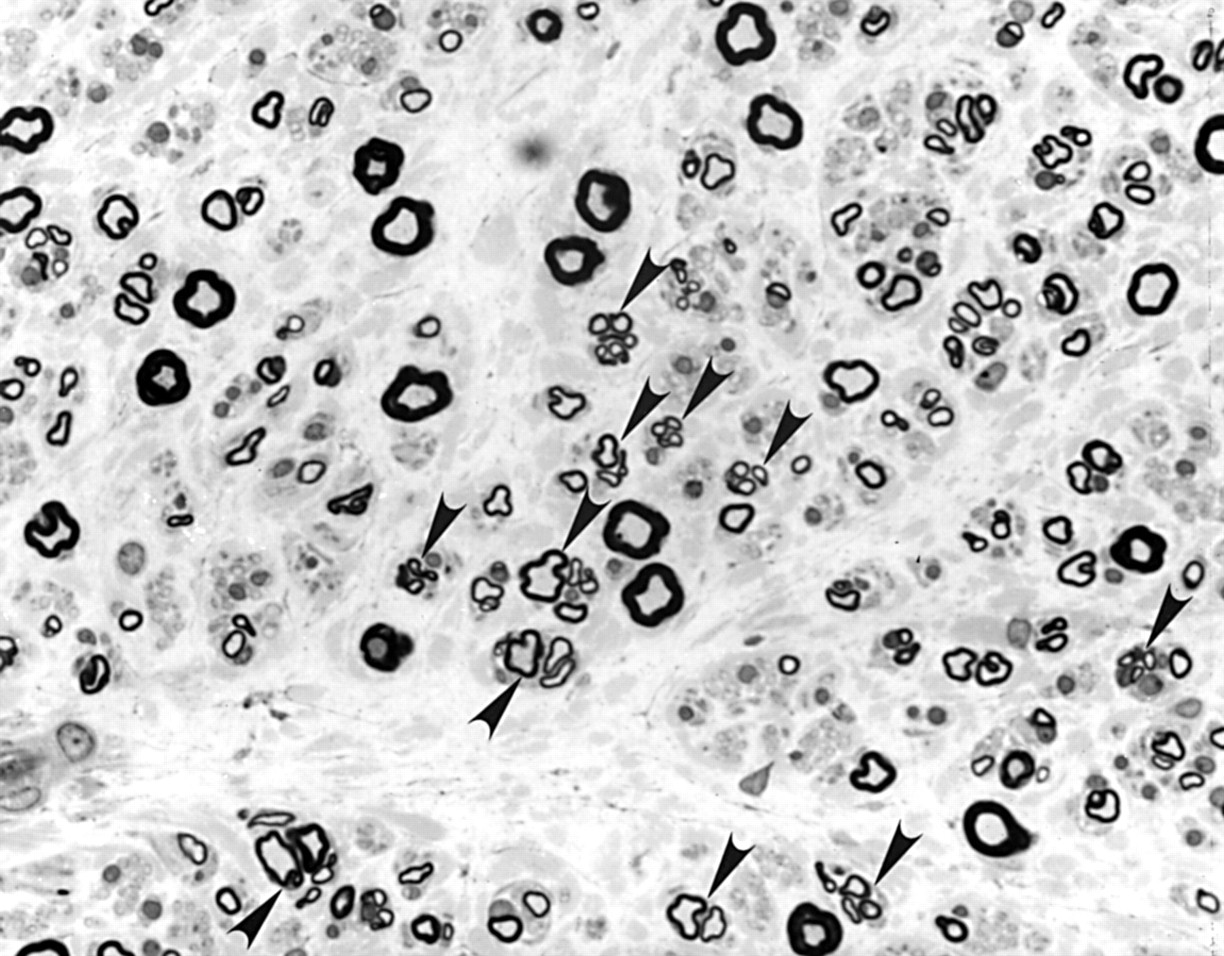
Sural nerve specimen obtained from proband from family 1. Toluidine blue stained transverse semithin sections (magnification×300) showing moderate oedema of the endoneural and subperineural space and a moderate myelinated fibre loss that was most pronounced in large fibres. Marked axonal sprouting (arrows) and atrophy were seen in residual myelinated fibre. Onion bulb formation was absent.

{kind=link}
{kind=link}
{kind=link}
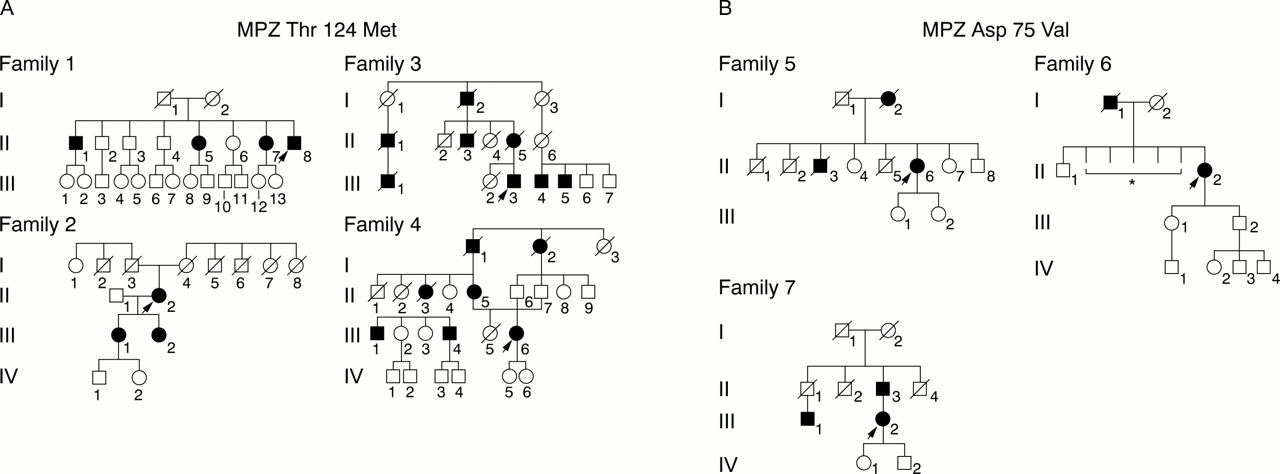
Pedigree of families with mutations in the peripheral myelin protein zero gene (Thr124Met or Asp75Val). (A) MPZ mutation Thr124Met. (B). MPZ mutation Asp75Val. Squares=males; circles=females; fully darkened symbols= affected males and females; diagonal lines=deceased; arrowheads=the proband's cases. *Detailed information was not available because they died during childhood.
Patient 6 (family 6)
In family 6 the proband was a 66 year old woman complaining of bilateral plantar paraesthesias that had gradually worsened since onset at the age of 61. The patient had shown normal physical abilities for age in elementary and middle school.
Neurological examination showed bilateral pupillary dilation and extremely slow light and convergence reflexes, with miosis in response to 0.125% pilocarpine; this was diagnosed as Adie's pupil. Auditory acuity was normal. Paraesthesias were noted in both legs. Pain, light touch, vibration, and joint position perception were reduced in both legs below the knees. Muscle strength seemed normal in the arms but was severely reduced distally in both legs. Amyotrophy of the calves and soles were seen. Toe walking could not be performed. Deep tendon reflexes were diffusely absent. No pathological reflexes were seen.
Concentration of creatine kinase was slightly increased in serum (376 IU/I). In CSF the protein concentration was normal (39 mg/dl). The median nerve MCV was 44 m/s and the CMAP was 6.5 mV, In the tibial nerve no CMAP could be evoked, The sural nerve exhibited an SCV of 41 m/s and a SNAP of 5.7 μV. The amplitudes of CMAPs and SNAPs were extremely reduced in the legs, but conduction velocity was well preserved. Analysis of DNA detected an A to T substitution at codon 75 in exon 2 of the MPZ gene, resulting in an amino acid change from Asp to Val (Asp75Val; fig 1 B a). The patient's father had a history of paraesthesias and amyotrophy in the legs since his 4th decade, but detailed information was not available.
CLINICAL FEATURES OF THE SEVEN FAMILIES
The clinical features of patients in the seven families are summarised in table 1. Major clinical features were indistinguishable between families with the Thr124Met mutation and those with the Asp75Val mutation (table 1). Initial symptoms occurred at 37 to 61 years (mean (SD) 50.4 (9.6), representing a relatively late onset. These symptoms were often paraesthesias or hypaesthesia involving the distal portion of the leg. Calf muscle cramps and weakness in the distal leg muscles were also seen as initial symptoms. In most patients, degree of sensory involvement was similar for superficial and deep sensory modalities. Limb muscle weakness and atrophy were noted to a mild or moderate degree in a distally accentuated manner, usually greatest in the legs. Ability to carry out activities of daily life was generally well preserved. Deep tendon reflexes were diminished or absent in all patients, especially in the legs. Deafness was present in five of the 11 patients, Adie's pupil in nine of the 13 patients. The serum creatine kinase concentration was slightly increased in six of the eight patients in whom it was examined.
Clinical features of patients in seven families with MPZ mutations
The nerve conduction velocity was well preserved (table 2). The CMAPs and SNAPs were decreased or not elicited, especially in the legs, which indicates axonal dysfunction. Three sural nerve biopsies were performed. All specimens showed moderate myelinated fibre loss and prominent axonal sprouting, whereas onion bulb formation was not evident in any specimen. Unmyelinated fibres showed an essentially normal density, but also showed sprouting. In teased fibre preparations, axonal change was predominant, but mild segmental demyelination and remyelination were seen in some patients. Characteristics of patients in the seven families included relatively late age of onset, relatively severe sensory involvement combined with some degree of motor impairment, and a frequent association with Adie's pupil, deafness, and increase in serum creatine kinase concentration.
Nerve conduction study in patients with an axonal form of CMT associated with a mutation in the MPZ
Discussion
Charcot-Marie-Tooth disease can be classified based on nerve conduction velocities into: CMT type 1 or demyelinating type shows a median nerve MCV of less than 38 m/s; CMT type 2 shows mainly axonal impairment while maintaining an MCV of 38 m/s or greater18; CMT1A and CMT1B present onion bulb formation and hypertrophy, which are pathological changes representative of demyelination.19 On the other hand, type 2 or axonal CMT includes many subgroups. Among these, gene locations for types 2A, 2B, 2D, and 2P are well known.1-3 20 The MPZ protein, which belongs to the globulin superfamily, is involved in cellular adhesion21 and is a main structural component of the myelin sheath. Mutations of the MPZ gene can cause the demyelinative disorder CMT1B, and also underlie Dejerine-Sottas disease or congenital hypomyelination.22 So far, more than 45 missense and nonsense mutations of the MPZ gene have been reported23; most of these produce the demyelinative clinical phenotype CMT type 1B. However, some CMT families with the MPZ gene mutation, showing an axonal phenotype with a relatively late onset, have been described in white populations.6-10 However, detailed clinical phenotypes, particularly those of associated symptoms, have not been documented in these mutations except for Thr124Met.7 8Our four Japanese families with Thr124Met mutation may support the view that this mutation is a “hot spot” of MPZ mutation among the different ethnic populations. Our families, with both Thr124Met or Asp75Val mutations in the MPZ gene, showed an axonal phenotype with only mild slowing of nerve conduction velocity but marked reduction of CMAP and SNAP amplitudes.
The most striking finding in these families was that the distinctive features were very similar between patients with Thr124Met and those with Asp75Val: relatively late onset, marked sensory impairment, and frequent association with Adie's pupil, deafness, and increased creatine kinase. The characteristics seen in families with Thr124Met were similar to those described by Jonghe et al 7 and by Chapon et al 8 in other families with the same mutation, confirming that the Thr124Met mutation in the MPZ gene causes a distinctive clinical phenotype including axonal neuropathy. Our similar findings in families with Asp75Val suggest that MPZ mutations causing axonal neuropathy are often associated with distinctive associated symptoms irrespective of the location and nature of the mutation in the MPZ gene.
Assessment of the MPZ gene should be included in the clinical diagnostic investigation of patients with CMT type 2. Furthermore, one patient in our series initially was misdiagnosed with Sjögren's syndrome because Adie's pupil was a prominent feature and sensory involvement showed a late onset,24 25 Patients showing Adie's pupil, deafness, or increased serum creatine kinase associated with a chronic neuropathy should undergo MPZ gene analysis. The molecular mechanisms that produce the phenotypes related to the different mutations in the MPZ gene are unknown. Common molecular pathways may be shared by axonal dysfunction and pupillary and auditory involvement in different MPZ mutations. Study of a transgenic mouse model with different mutations of the MPZ gene should help to clarify this issue.