Article Text

Abstract
Linear scleroderma “en coup de sabre” (LScs) is associated with neurological complications, the pathogenesis of which is uncertain. A 27 year old woman is reported on who developed epilepsy and focal neurological signs in association with LScs. Brain MRI demonstrated predominantly ipsilateral relapsing and remitting grey and white matter lesions. Analysis of CSF and pathology obtained at brain biopsy provides evidence of an inflammatory process which may be amenable to immunosuppressive treatment.
- scleroderma “en coup de sabre”
- central nervous system inflammation
- progressive facial hemiatrophy
Statistics from Altmetric.com
Linear scleroderma “en coup de sabre” (LScs) denotes linear scleroderma of the frontoparietal area of the head. It has been reported in association with ipsilateral intracerebral lesions, epilepsy, and ocular complications. Ipsilateral white matter lesions have been seen on CT and MRI in patients with LScs,1-5 but there are only two reports of neuropathology.4 5 We report a unique case of LScs where intracerebral lesions which correlate with the clinical course have arisen and resolved spontaneously as recorded with MRI. Neuropathological findings, intrathecal production of IgG, and the response to immunosuppressive treatment, provide evidence for a potentially treatable primary intracerebral inflammatory process occurring in association with linear scleroderma. We discuss these findings in relation to the overlapping and sometimes coexistent condition of progressive facial hemiatrophy (PFHA, also named Parry-Romberg syndrome) in which characteristically the lower half of the face becomes unilaterally atrophic.
Case report
In 1989 a 27 year old right handed woman presented with a 3 month history of weakness in her left hand. Aged 15 she had presented with typical LScs affecting the right frontotemporal area of her scalp. This had subsequently progressed over several years but had been static for 5 years. She had been under regular dermatology review and received no treatment. There was no other significant medical or family history. On examination, there was mild pyramidal weakness of the left arm with exaggerated tendon reflexes. There were no other manifestations of connective tissue disease.
Brain MRI initially showed extensive abnormalities in the right hemisphere comprising multiple high signal foci on T2 weighted images involving grey matter, and superficial and deep white matter. There was enhancement with gadolinium on T1 weighted images and mild focal atrophy. The constituents of CSF were normal but electrophoresis showed local synthesis of IgG with an oligoclonal distribution.
Six months later she presented with secondarily generalised seizures. A repeat MRI examination showed partial resolution of the previous lesions but a new area of high signal in the right frontal lobe. Routine laboratory studies and a comprehensive autoantibody screen were negative.
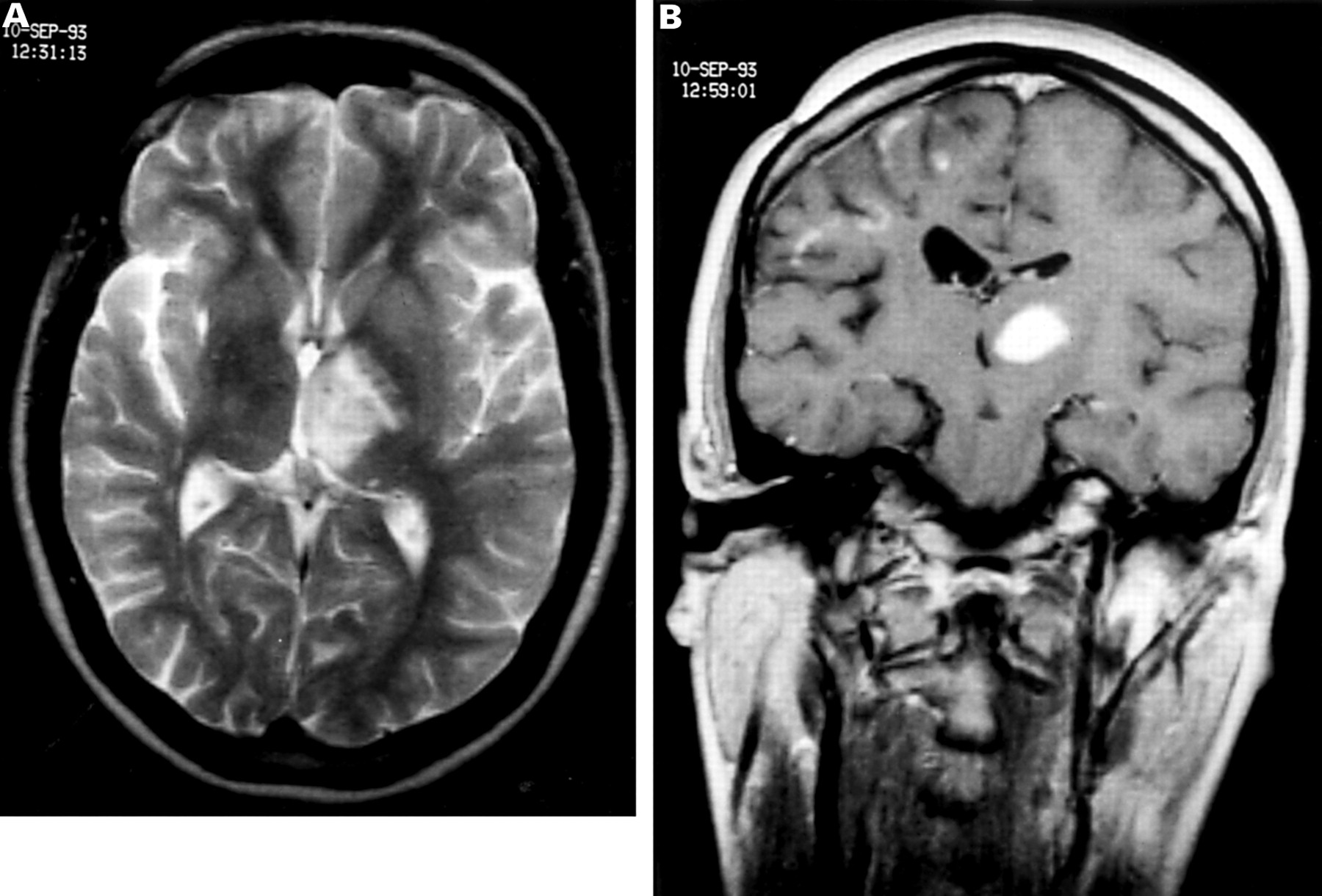
Over the next 3 years her epilepsy remained well controlled on anticonvulsant drugs. Brain MRI in 1992 showed partial resolution of the previous right hemispheric lesions. In 1993 she began to experience difficulties with handwriting, dysarthria, and jaw spasm. Repeat MRI demonstrated a large new high signal lesion on T2 weighted images in the left thalamus, with marked enhancement on T1 weighted images and a new lesion in the cerebellum (fig 1 ).Because of concern that this might be neoplastic, an uncomplicated stereotactic biopsy of the left thalamic lesion was performed.

MRI in 1993 before biopsy. (A) Axial T2 weighted fast spin echo image showing an area of high signal predominantly involving the left thalamus with mild mass effect. There are several other small foci of high signal involving the white matter adjacent to the trigone of the right ventricle and deep to the insular cortex. (B) Coronal gadolinium enhanced T1 weighted spin echo image demonstrating the left thalamic lesion, with marked uniform enhancement, distorting the third and left lateral ventricles. There are several foci of enhancement involving grey matter within the right frontal lobe and atrophy of the white matter with dilatation of the right lateral ventricle. The scleroderma “en coup de sabre” is seen with focal atrophy of the subcutaneous tissues on the right.
Histology showed a focal inflammatory process centred on blood vessels with infiltration but not necrosis of the vessel walls. Lymphocytes and monocytes infiltrated the adjacent brain, which showed astrocytic enlargement, and in one area foamy macrophages in the neuropil merged with a zone of necrosis. In places lymphocytes surrounded single necrotic neurons. Stains for bacteria, protozoa, and fungi were all negative. Polymerase chain reaction (PCR) based techniques to detect herpes simplex virus, measles, and Epstein-Barr virus were negative. The appearances were interpreted as a low grade vasculitis with associated focal cerebral necrosis (fig 2).

Biopsy of thalamic lesion. (A) Small vessels show patchy perivascular lymphocytic infiltrate which extends into adjacent brain. Astrocytic hypertrophy is evident and in the top right, foamy macrophages indicate focal necrosis (haematoxylin and eosin originally×280 magnification). (B) A more intensely inflamed vessel with a predominantly lymphocytic infiltrate at the edge of an area of necrosis (haematoxylin and eosin×280 magnification).
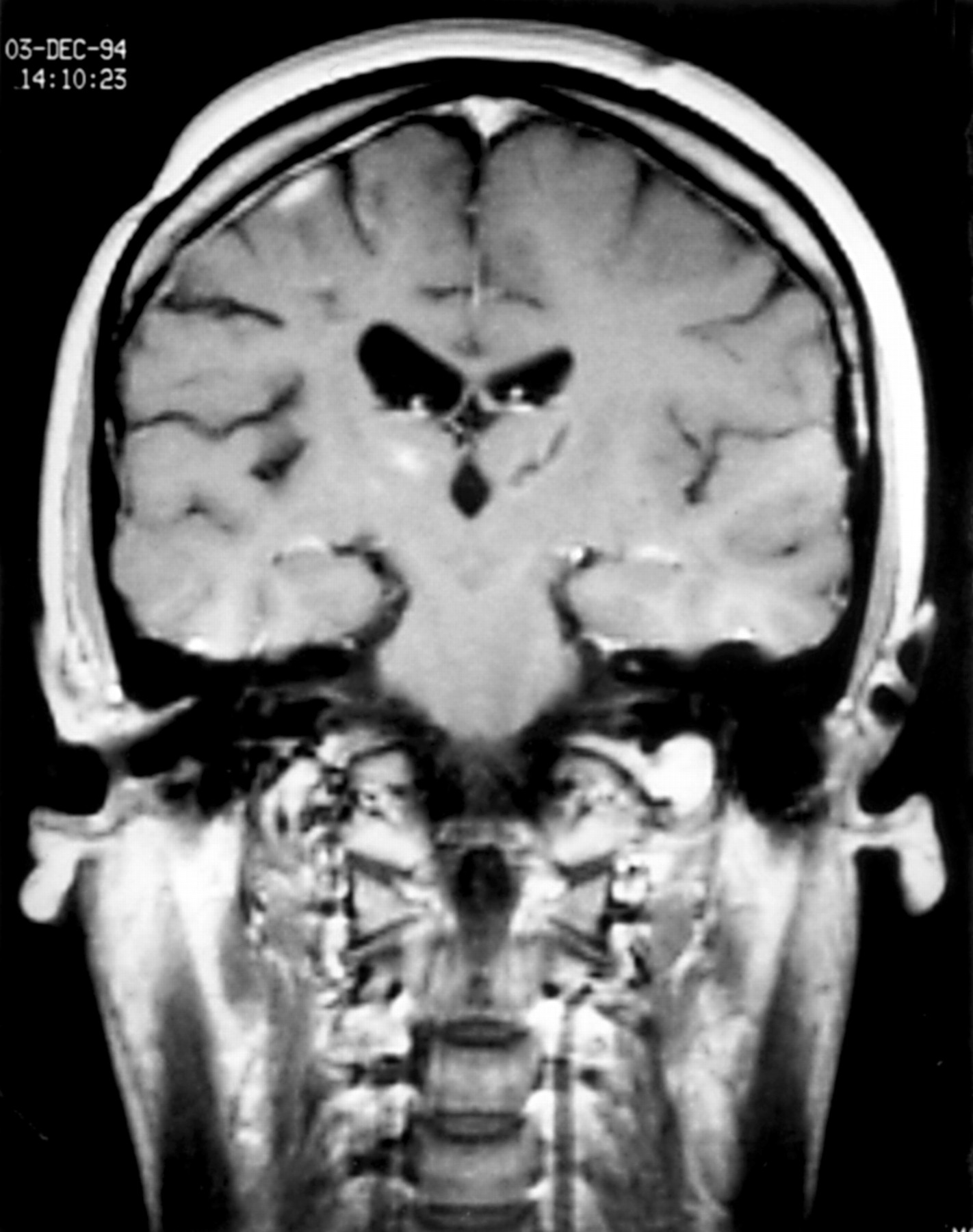
Our patient again improved spontaneously until 1994 when she returned with further secondarily generalised seizures. Brain MRI showed significant resolution of the left thalamic lesion with new areas of high signal predominantly in the right frontal lobe (fig 3). Further CSF examination showed 12 lymphocytes/mm3 with normal protein and glucose. She continued to have seizures and developed a mild Raynaud's phenomenon in her left hand. She was treated with pulsed methylprednisolone (1g daily for 3 days). A repeat MRI displayed some resolution of the lesions and her clinical condition improved. In 1996, after there had been deterioration in the function of her left hand, she was treated with intravenous cyclophosphamide. After a pulse of treatment, she considered that she was much improved and was able to extend the fingers of her left hand. She therefore started azathioprine in March 1996. She was unable to take more than 100 mg daily because of lymphopenia. An MRI in Jan 1997 showed no significant change except that the overall extent of the enhancement had reduced. By May 1997 she was able to straighten her fingers but not her thumb. She was still having fits about once a month. A scan in July 1997 showed little change except that there was less enhancement than in 1994. During 1998 her fingers became more flexed and her seizures have continued at about the same interval since. The linear scleroderma is static and there is no evidence of progression to facial hemiatrophy.
{kind=link}
{kind=link}
{kind=link}
Brain MRI in 1994 1 year after thalamic biopsy. Coronal gadolinium enhanced T1 weighted image showing the site of the biopsy as a linear low signal in the left thalamus. The enhancement and mass effect has resolved. The pattern of enhancement within the right frontal lobe has changed and there is a new lesion within the right thalamus.
Discussion
Linear scleroderma “en coup de sabre”, its relation with PFHA, and their neurological complications have been discussed by authors since the 19th century. Both have been reported in association with focal contralateral seixures and pyramidal signs, ipsilateral intracranial calcification, and ophthalmological abnormalities. Although a distinction can be drawn between the atrophy of the deeper tissues in PFHA and the more prominent skin induration in LScs,3 6 they are almost certainly overlapping conditions and sometimes coexist. The close nature of these two conditions means that they should be discussed in parallel.
There are several reports of MRI findings in patients with LScs. Hyperintense, predominantly ipsilateral, cortical and subcortical lesions seen on MRI were described in a 32 year old woman who also had uveitis, Raynaud's phenomenon, and intrathecal synthesis of IgG.1 This case demonstrated some spontaneous remission of lesions. Liu et al reported ipsilateral changes in two children, consisting of calcification and abnormal grey and white matter interpreted as a migrational abnormality and dysmyelination.2 Chung et al 4 have reported on a 27 year old person with a large ipsilateral calcified cortical and subcortical lesion. Our patient is unusual in showing relapsing and remitting lesions that are not all confined to the same side as the skin lesion. Coincidental multiple sclerosis is unlikely given the involvement of grey and white matter.
Neuropathology in LScs has been reported in only two patients. Chunget al 4 found leptomeningeal band-like sclerosis, intraparenchymal calcification, and ectatic vessels with gliosis but no evidence of inflammation. Dubeauet al 5 reported chronic inflammatory changes with perivascular infiltrates, neuronal loss, and gliosis similar to our patient. Further evidence for an inflammatory basis to both LScs and PFHA comes from serological studies which show high frequencies of positive serum antinuclear antibodies and rheumatoid factor.7 8
The neuroradiological and pathological correlates of PFHA are similar. Fry et al 9 reported on five children with upper facial hemiatrophy who had high signal white matter lesions seen on MRI, predominantly in the frontal lobe (four ipsilateral, one contralateral). Terstegge et al 10 reviewed most of the available radiological evidence and reported on a similar patient. They also commented on the presence of ill defined sulci in the presence of ventricular dilatation, suggesting meningeal adhesions. Dupontet al 11 reported on four patients with parasagittal PFHA who had focal MRI changes consisting of ipsilateral cortical thickening, gyral enhancement, and blurring of the grey-white interface. Hyperintense lesions on T2 weighted sequences were also seen. Cory et al 12described a child with similar but more severe findings including changes interpreted as unilateral focal infarction in the corpus callosum, diffuse white matter changes, and leptomeningeal enhancement. Goldberg-Stern et al described a child who developed facial hemiatrophy with uveitis and epilepsy. Brain MRI disclosed ipsilateral meningeal and basal ganglia lesions similar to those in our patient. Their patient's lesions and ophthalmological problems seemed to resolve after starting methotrexate.13There are case reports of angiographic evidence of ipsilateral reversible vessel calibre changes,14 bilateral intracranial aneurysms,15 carotid dissection,16 and arteriovenous malformation.6 There have been five reports of intracranial neuropathology in PFHA since the condition was first described in 1823. Two early German reports17 18commented on thickened opaque meninges in association with meningeal neovascularisation and atrophic underlying cortex. Eadieet al 19 and Merrittet al 20 commented only on cerebrocerebellar calcification and ventricular dilatation. Wolf and Verity21 also found leptomeningeal fibrosis, degenerative cortical changes, and atypical small tortuous meningeal vessels interpreted as a microvascular malformation. Wartenberg22described ipsilateral grey and white matter perivascular lymphocytic infiltration with marked pial and glial proliferation.
The dominant picture in this cerebral biopsy is of an inflammatory process involving blood vessels and resulting in focal cerebral necrosis. This is in keeping with the intense inflammation of the dermis and subcutaneous tissue seen in the early stages of LScs. We propose that this case, as with the case of Dubeau et al,5 supports the hypothesis that there is an early cerebral inflammatory stage in LScs which can later progress to the end stage pathology found by Chunget al.4 This challenges their conclusions that the process is developmental and is supported by analogous neuropathological findings in PFHA. The oligoclonal response in the CSF, and the response to immunosuppression in our patient indicate that there is an inflammatory or immune mediated mechanism underlying the associated cerebral process in LScs and that treatment should be considered when clinically relevant.
Intracerebral abnormality should always be considered in patients with localised scleroderma of the head or PFHA. The investigation of choice is MRI and this may identify further asymptomatic cases. The similarity of intracranial manifestations in these two disorders suggests that they share a similar aetiology and are probably overlapping syndromes.
Acknowledgments
Consent has been obtained from the patient for the publication of this case report.