Article Text

Abstract
Objectives: To predict the size of the vCJD epidemic it is important to know whether the description of cases of vCJD in 1996 represent the first cases of a new disease entity or whether detection was due to increased surveillance of CJD in humans. Detection of earlier cases would suggest a shorter incubation period and might lead to predictions of epidemic size being revised.
Methods: All certified deaths (excluding external injury and poisoning) in residents of Wales aged 15–45, between 1985 and 1995, were reviewed to detect vCJD deaths that might have been overlooked. 12 091 deaths were reviewed. “Non-specific fatal disorders compatible with vCJD” were defined. Deaths recorded to diseases other than those defined were rejected from further analysis (8769). Remaining cases (3322) were subdivided. Group A comprised deaths recorded to suicide, transport accidents, and those that could not be ascertained (ICD9 rubrics E950–959, E800–848, and 7999), a total of 2698 cases. Group B comprised deaths due to neurological disease, psychiatric disease, or substance abuse (624).
Results: For group A, remaining brain material was identified (n=218, 8.1%) and examined by routine histology and immunocytochemistry for prion protein. No cases of vCJD were detected. For group B, review of remaining clinical information was undertaken. Of 624 cases, information was available on 447 (72%). Brain tissue was examined by routine histology and immunocytochemistry in 47 (7.5%) cases. Sufficient clinical and pathological information was available to exclude all these as potential cases of vCJD.
Conclusion: Variant CJD is a new disease entity and not simply the result of better case ascertainment.
- variant Creutzfeldt-Jakob disease
- epidemiology
- prion protein
- immunocytochemistry
- NCJDSU, National Creutzfeldt-Jakob disease Surveillance Unit
- ICD-9, ninth revision of the international classification of diseases
- ONS, Office of National Statistics
- E codes, external causes of death
- BSE, bovine spongiform encephalitis
- Prp, prion protein
Statistics from Altmetric.com
- NCJDSU, National Creutzfeldt-Jakob disease Surveillance Unit
- ICD-9, ninth revision of the international classification of diseases
- ONS, Office of National Statistics
- E codes, external causes of death
- BSE, bovine spongiform encephalitis
- Prp, prion protein
The description of 10 cases of variant Creutzfeldt-Jakob disease (vCJD) in 19961 has already had enormous impact on public health and economic wellbeing. Strong evidence exists that vCJD is caused by infection with the same agent that causes bovine spongiform encephalitis (BSE).2–5 It is important to know, therefore, whether cases may have occurred in the past and whether the recognition of the first cases between 1994 and 1995 represents a new disease entity or is merely the result of better case ascertainment. The authors from the National Creutzfeldt-Jakob disease Surveillance Unit (NCJDSU) think that vCJD is a new disease entity. The question, however, requires systematic examination.
It is vitally important to know, in a defined population, the incidence of diagnoses that could be compatible, even broadly, with vCJD. This would quantify the upper limit of the number of cases and identify the range of diagnoses within which vCJD might have been overlooked in the past and indicate the scope now for an artefactual epidemic. That is to say; has vCJD been endemic in the population, albeit in small numbers, but merely been miscoded under other diagnostic rubrics such as for example, multiple sclerosis, or epilepsy? Both questions are of concern to policy makers. It would also provide a baseline for future surveillance and indicate whether the incidence of diagnoses has increased over the study period.
METHODS
Ethical approval was sought from all Wales local research ethics committees. Death certificate data were requested from the Office of National Statistics (ONS) on deaths occurring in those aged 15–45 who were resident in Wales at the time of their death between the beginning of 1985 and the end of 1995 excluding those ascribed to International Classification of Diseases version 9 (ICD-9) rubrics 800–999 (injury and poisoning). External causes of death (E codes) were also excluded except for the rubrics E800–848 (transport accidents) and E950–959 (suicide and self inflicted injury).
A steering committee was set up to decide a priori which ICD-9 diagnoses might be compatible with a diagnosis of vCJD. A list of ICD-9 codes entitled “non-specific fatal disorders compatible with vCJD” was compiled (see box).
Each death certificate was then reviewed manually and either accepted into the “non-specific fatal disorders compatible with vCJD” group or excluded from further analysis.
The group “non-specific fatal disorders compatible with vCJD” was further divided into two separate groups: group A and group B. Group A comprised deaths recorded to rubrics for suicide, transport accidents, and those deaths that could not be ascertained (ICD9 rubrics E950–959, E800–848, and 7999). Unascertained causes of death were cases in which even after a postmortem examination the pathologist could not be sure of the underlying cause of death. Group B comprised deaths due to neurological disease, psychiatric disorder (other than suicide), or substance abuse. In the case of group A (suicide, transport accidents, and unascertainable causes of death), existing brain material was traced and examined. In the case of group B all existing clinical information was traced and reviewed. After this, existing brain material was traced and examined in any case who had an illness duration of 3 years or less (see fig 1). After all available clinical data had been collected for group B, three sets of selection criteria were applied. Firstly, cases with duration of illness exceeding 3 years were excluded. Secondly, cases with definitive clinical, laboratory, or pathological evidence of another diagnosis were excluded. Thirdly, cases in which activity just before death was incompatible with a rapidly evolving dementia were excluded.

Methods and results.
ICD-9 rubrics: Non-specific fatal disorders compatible with vCJD
054 Herpes simplex.
046 Slow virus infection of the CNS (includesKuru,Jakob-Creutzfeldt disease,subacute sclerosing panencephalitis, progressive multifocal leucoencephalopathy, other, unspecified).
265 Thiamine and niacin deficiency states (includespellagra).
266 Deficiency of B complex components.
290 Senile and presenile organic and psychotic conditions (includessenile dementia simple type, presenile dementia, senile dementia depressed or paranoid type, senile dementia with acute confusional state, arteriosclerotic dementia, other, unspecified).
291 Alcoholic psychoses (includesdelirium tremens.Korsakov's psychosis, alcoholic,other alcoholic dementia).
292 Drug psychoses.
293 Transient organic psychotic conditions (includesacute confusional state,subacute confusional state).
294 Other organic psychotic conditions -chronic (includesKorsakov's psychosis or syndrome (non-alcoholic),dementia in conditions classified elsewhere).
295 Schizophrenic psychoses.
296 Affective psychoses.
297 Paranoid states.
298 Other non-organic psychoses (includesunspecified psychosis).
299 Psychoses with origin specific to childhood.
300 Neurotic disorders (includesanxiety states, hysteria, phobic state, obsessive-compulsive disorders, neurotic depression,neurathenia,depersonalisation syndrome,hypochondriasis, other neurotic disorders,unspecified).
301 Personality disorders.
302 Sexual deviations and disorders.
303 Alcohol dependence syndrome.
304 Drug dependence.
305 Non-dependent abuse of drugs.
306 Physiological malfunction arising from mental factors.
307 Special symptoms or syndromes not elsewhere classified.
308 Acute reaction to stress.
309 Adjustment reaction.
310 Specific non-psychotic mental disorders following organic brain damage (includesfrontal lobe syndrome).
311 Depressive disorder, not elsewhere classified.
312 Disturbance of conduct not elsewhere classified.
313 Disturbances of emotions specific to childhood and adolescence (includes with anxiety and fearfulness).
323 Encephalitis, myelitis and encephalomyelitis (includespoliomyelitis, athropod-borne viral encephalitis, other encephalitis due to infection,encephalitis following immunisation procedures,postinfectious encephalitis,toxic encephalitis,other, unspecified cause).
330 Cerebral degenerations usually manifest in childhood (includescerebral lipodoses, cerebral degeneration in generalised lipidoses,cerebral degeneration of childhood and other diseases classified elsewhere, other cerebral degenerations in childhood ,unspecified).
331 Other cerebral degenerations (includesAlzheimer's disease,Pick's disease,senile degeneration of brain, communicating hydrocephalus, obstructive hydrocephalus,cerebral degeneration in other diseases classified elsewhere,other cerebral degenerations,unspecified).
332 Parkinson's disease.
333 Other extrapyramidal disease and abnormal movement disorders (includesother degenerative diseases of the basal ganglia, essential and other specified forms of tremor,myoclonus, tics of organic origin,Huntington's chorea,other choreas, idiopathic torsion dystonia,symptomatic torsion dystonia, fragments of torsion dystonia,other and unspecified).
334 Spinocerebellar disease (includesFriedriech's ataxia, Hereditary spastic paraplegia,primary cerebellar degeneration,other cerebellar ataxia,cerebellar ataxia in disease classified, elsewhere,other, unspecified).
335 Anterior horn cell disease.
337 Disorders of the autonomic nervous system.
340 Multiple sclerosis.
345 Epilepsy.
507 Pneumonitis due to solids liquids (includes aspiration pneumonia).
579 Intestinal malabsorption (includescoeliac disease).
742 Other congenital anomalies of nervous system (includes 742.8other unspecified anomalies of nervous system).
780 General symptoms (includesconvulsions).
781 Symptoms involving nervous and musculoskeletal systems (includesabnormal involuntary movements,abnormality of gait,lack of coordination).
799 Other ill-defined and unknown causes of morbidity and mortality (includesasphyxia,respiratory failure, nervousness, debility-unspecified,cachexia, other ill defined conditions,other unknown and unspecified cause).
E800-E848 Transport accidents.
E950-E959 Suicide and self inflicted injury.
Each NHS trust in Wales was asked for permission to access hospital notes of those patients in the “neurological, other psychiatric disease or substance abuse” group who had died in hospital in that trust. In addition, each of the health authorities in Wales were approached with a request to view existing general practitioner records and all coroners involved in the deaths of residents of Wales were asked for permission to view coroners' files. Relevant data obtained was entered into a standardised proforma.
Pathologists in Wales and those responsible for postmortem examinations of Welsh residents in England were contacted and asked to provide existing samples of cerebral or spinal cord tissue on cases in the “non-specific fatal disorders compatible with vCJD”.
Blocks for routine histological assessment were prepared using a haematoxylin and eosin stain. In most cases a block of cerebral cortex was used for immunocytochemistry; however, in some cases blocks of cerebellum and brain stem were also examined. Findings from the most pathologically abnormal part of the section were recorded. An arbitrary scale of hypoxic induced change was adopted. The presence of spongiform change, plaques, and/or gliosis was recorded.
Immunocytochemistry was performed on the same paraffin embedded tissue blocks examined by routine histology; 4 μm sections were cut from tissue blocks and pretreated by autoclaving at 121°C for 10 minutes followed by immersion in 98% formic acid for 5 minutes. Prion protein (PrP)was detected by the well characterised and widely used monoclonal antibody KG9 (IAH TSE Resource Centre, Compston, UK) at a dilution of 1:200 overnight and visualised with the Elite universal detection kit (Vector, UK) and colour was developed with diamino benzidine.6,7 A positive control of vCJD cerebral tissue was included. The absence or presence of PrP staining was recorded together with the pattern of staining seen. The project neuropathologists were blind to patient details.
RESULTS
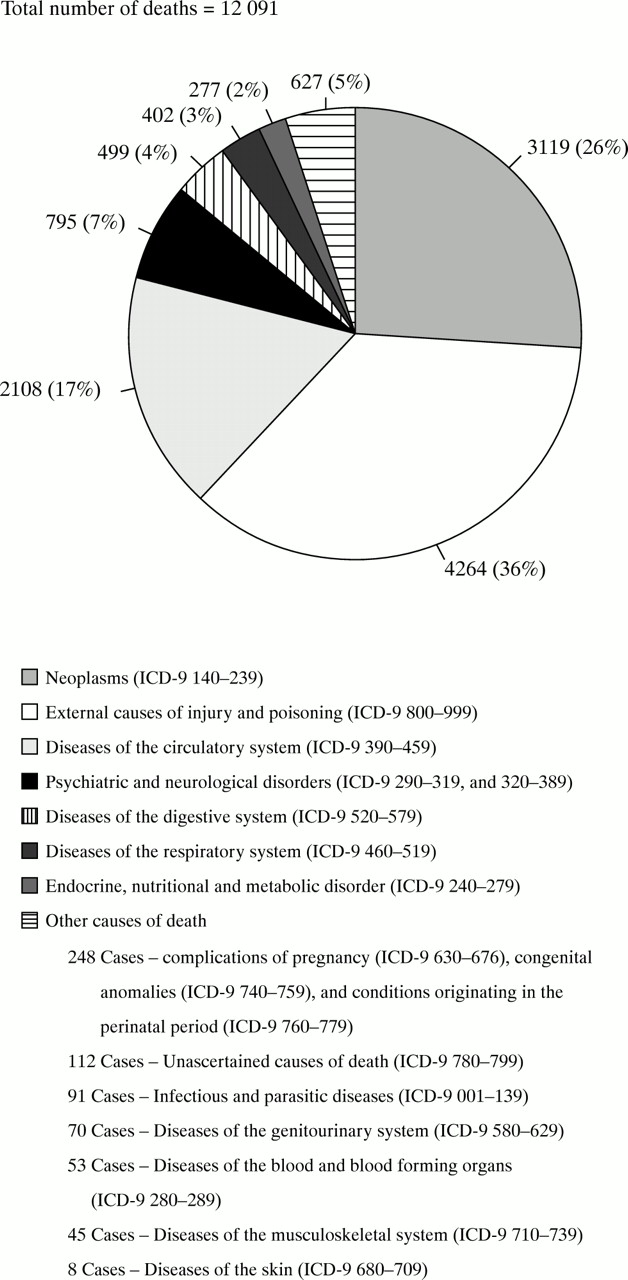
For the period 1985–95 there were 12 091 deaths (excluding external injury and poisoning). Figure 2 shows the proportion of deaths attributed to each of the ICD9 major diagnostic categories for the 12 091 deaths.
{kind=link}
{kind=link}
Proportions of deaths attributed to each of the ICD-9 major diagnostic categories.
There were 3322 deaths in the “non-specific fatal disorders compatible with vCJD” group. Group A comprised deaths coded to suicide (n=1285), transport accident (n=1309), and unascertained causes (n=104). Group B consisted of all other ICD9 rubrics in the “non-specific fatal neurological disorders compatible with vCJD” group, a total of 624 cases (see table 1).
Demographic and clinical details of subjects in group B
Paraffin brain blocks were retrieved for 218 (8.1%) cases included in group A. Of these, 116 brain blocks were retrieved from transport accident cases (8.8% of the transport accident group), 88 blocks from suicide deaths (6.8% of the suicide deaths), and 14 blocks from the “unascertained” group (13.4% of unascertained group). Some hypoxic change was seen in 60% of cases. Severe hypoxic change correlated with transport deaths and reflected the frequency of severe head injury in this group. Gliosis was seen in 12 cases. Vacuolation was seen in 29 cases but judged to be non-specific vacuolation involving the cortex resulting from either brain oedema, artefacts resulting from histological processing and/or recent hypoxia/ ischaemia. No pathological plaques were seen in any of the samples.
Overall, data were available for 447 of the 624 cases in group B (72%). Of the 267 requested hospital records, 130 (49%) were available for review. General practitioner records were requested on 622 of the 624 cases (two were unable to be traced due to no fixed abode), 143 (23%) were available for review. Coroners' files were requested on 384 of the 624 cases, and 308 (80%) were available for review. Pathological material was retrieved from 16 of the 17 pathology departments approached in Wales. Some pathological material was retrieved from one of 11 pathology departments approached in England (a total of 21 cases). Among those it was necessary to exclude due to there being no further information included three cases with presenile dementia written on the death certificate. The first case, a 36 year old woman, died in 1988, the death certificate also included “cerebral lupus erythematosus”. In the other two cases, the final illness was recorded as bronchopneumonia, in one case, a man aged 44 died in 1988 in a community hospital and the other died in a long stay hospital in 1987, aged 45.
Of the 624 patients in group B, 407 (65%) were known to have undergone postmortem from information recorded on death certificates. Detailed information was available on 330 of 407 patients of which 135 patients had a clinical history of less than 3 years or not enough information was available to confirm the duration of illness (the last were included). Brain tissue was retrieved on 47 (35%) of these cases. Some hypoxic change was seen in 24 of the 47 cases. Severe hypoxic change was seen in only four cases. Gliosis was seen in two cases. Vacuolation was seen in five cases but judged to be non-specific vacuolation involving the cortex resulting from either brain oedema, artefacts resulting from histological processing, and/or recent hypoxia/ ischaemia. No spongiform change or pathological plaques were seen in any of the samples.
Immunocytochemistry was performed on all blocks from cases in group A and group B, a minimum of one block/case. None of the cases showed patterns of PrP immunoreactivity seen in vCJD. Forty seven per cent of cases showed some weak PrP immunoreactivity predominantly within neurons, but occasionally axons. The neuronal staining corresponded with acute hypoxic/ischaemic neuronal change on haematoxylin and eosin stained sections whereas the axonal staining corresponded to either potential foci of diffuse axonal injury in the white matter or was seen adjacent to an area of cortical infarction.
Of 447 cases in which some clinical information was available, 283 (63%) were excluded as illness duration was recorded as being 3 years or longer: 160 (38%) with duration of illness of less than 3 years were excluded because there was clear clinical, laboratory, or pathological evidence for an alternative diagnosis to vCJD. Finally, five cases were excluded on the basis that, despite insufficient clinical information being available to confirm a diagnosis other than vCJD, activity found before death was incompatible with an established dementia. All five cases were certified as having died of epilepsy. In four cases the person was known to have had epilepsy but duration of illness was not stated in the records retrieved. In each of these cases the deceased was seen to have been acting normally hours before death as judged by a close relative, and in one case by the deceased's landlady. In the final case the deceased had been fit and well until 10 days before his admission to hospital.
No case met the CJDSU criteria for even possible vCJD.8
Assuming a Poisson distribution for vCJD, the upper 95% confidence interval for an observed occurrence of zero cases would be 3.7 cases during the study period. The average population of Wales for all age groups (per annum) during the study period was 2 873 348. Assuming that the only cases of vCJD would have occurred between the ages of 15–45, the upper 95% confidence interval for the incidence of death for vCJD/million/annum was 0.12.
DISCUSSION
The decision to review death certificates for the 11 years preceding the first descriptions of vCJD1 was based on the veterinary data suggesting that BSE was a new disease that emerged in the early 1980s.9 Given that the favoured explanation for the emergence of vCJD was that it was due to the exposure of BSE infected material, experience gained from studying other human transmissible spongiform encephalopathies suggested the incubation period in vCJD was likely to be measured in years. If this study had detected cases of vCJD as far back as 1985, such a finding would have cast doubt over the proposed link between BSE and vCJD. Indeed there may have been an argument for extending the study period back to 1980 when, if a case had been discovered, one could be reasonably confident that vCJD was not caused by direct exposure to BSE. In retrospect, however, any extension of the study back to 1980 would have resulted in very little clinical information being available for review due to the unavailability of clinical notes owing to current destruction policies.
The chosen age range of 15–45 was proposed after the description of the first 10 cases of vCJD, where age of the patients ranged from 19–41 years.1 Up to the end of 1999 there had been 52 confirmed cases of vCJD10 of which only four patients were aged over the age of 50 (7.6%). No patient died of vCJD below the age of 15. Based on these figures we think that the conclusions drawn by this paper still remain valid.
A steering committee was appointed to produce a list of ICD9 rubrics entitled “non-specific fatal disorders compatible with vCJD”. Each member of the appointed committee was made familiar with the published clinical phenotype of the vCJD cases and then provided with a list of ICD9 rubrics on which they were asked to mark down any diagnosis that might be mistaken for vCJD at any point in the clinical illness. Included in the list were ICD9 rubrics used by the NCJDSU11 and those used in a recent retrospective case note review of deaths from dementia in England.12
A comprehensive range of psychiatric rubrics was included to reflect the wide range of psychiatric presenting symptoms reported in vCJD.13 All deaths coded to rubrics for substance abuse (including alcohol) were included in an attempt to determine if substance abuse might be an early marker for the psychiatric prodrome of vCJD.
After the initial selection process the number of cases in the “non-specific fatal disorders compatible with vCJD” group totalled 3322. The decision to further subdivide this group into A and B was based on the assumption that there would be little or no relevant clinical information available on cases of transport accident deaths, suicides, and cases of death with no apparent cause and that a focused search for relevant clinical information in the neurological, psychiatric, and substance abuse group was more likely to unveil undiagnosed cases of vCJD.
The examination of just one brain block per case in most cases is, perhaps, a shortcoming of this study. Extensive sampling from various brain areas (minimum from frontal, temporal, and occipital lobes, basal ganglia, and cerebellum) is recommended in every postmortem of suspected spongiform encephalopathy.14 In this study, in none of the cases reviewed were the patients suspected to have a spongiform encephalopathy at the time of their death and this explains the limited numbers of brain blocks taken and rarity with which cases were referred for formal neuropathological review. However, one block of tissue with typical histopathology and/or unambiguous PrP immunoreactivity would provide a definitive diagnosis.14 It is encouraging that the pathological changes associated with vCJD are seen throughout the brain and this is particularly noted after PrP immunocytochemistry.15 Thus it is less likely that cases of vCJD were missed through limited sampling alone.
It is of importance that 47% of cases showed some weak PrP immunoreactivity with the KG9 mouse antibody. This finding in non-vCJD cases has been described in detail by Esiri et al.16 Although the incidental PrP staining seen both here and in the study by Esiri et al differs from that in vCJD and other forms of human spongiform encephalopathies, neuropathologists need to be aware of its appearance when analysing retrospective cases of undiagnosed dementia as possible cases of prion disease.16
Clearly, not all 447 patients of the 624 in group B could have had vCJD. Three exclusion criteria were applied in an attempt to produce a list of cases that would be compatible with vCJD.
The first criterion was based on the duration of clinical illness. In the first two detailed reports on clinical features of vCJD published on the first 10 and 14 cases respectively1,17 the maximum duration of illness from onset of symptoms to death was 35 months. Based on this information an exclusion criterion of illness of longer than 36 months in duration was applied. Was this a valid assumption? In the 52 cases recorded up to the end of 1999 only one case was recorded as having illness duration of over 3 years (38 months).10
The second selection criterion assumed that if there was clinical, laboratory, and/or pathological evidence for the disease recorded on the death certificate, it was unlikely that the patient also had vCJD. It is important to establish clear operational definitions for diseases as for other variables. In a retrospective study relying on case notes, this is far from an easy task. Outside of formal studies such as clinical trials, use is rarely made of rigid diagnostic rules. Diagnosis by clinicians tends to be based on a conceptual rather than on an explicit operational definition. For this study we used the operational diagnosis of the attending physician and were therefore tied to the accuracy of the original diagnosis. However, some degree of latitude was allowed in order to question the original diagnosis if any case met criteria for even possible vCJD, as defined by NCJDSU.8 The third selection criterion, “activity considered incompatible with established dementia” was not defined prospectively. Instead a pragmatic judgement was made in each of the five cases and on this basis it was judged that vCJD was unlikely. However, what activity is incompatible with dementia? As Will et al point out, the diagnosis of early symptomatic vCJD on clinical ground alone is virtually impossible.13
The finding of no cases of vCJD in the study population, assuming that the only cases of vCJD would have occurred between the ages of 15–45 during the study period, gives the upper 95% confidence interval for the incidence of death from vCJD in Wales as 0.12 per million/annum. If vCJD is, in truth, the result of better case ascertainment, rather than a new disease, then it could be argued that incidence of vCJD per million/annum should have been the same for 1985–94 as for 1995–2000. The observed mortality rate/ million/ annum for 1995–2000 in Wales was 0.29.10 This is two and a half times higher than the upper 95% confidence interval for the incidence of zero found in this study. This indicates an increase in incidence in the past 5 years which is unlikely to be due to chance alone. It suggests that vCJD is indeed a new disease.
Alternatively it could be proposed that vCJD has been endemic in the population, at albeit low levels, for many years and cases between 1985 and 1994 were missed by the study. If this study stood alone as the only piece of epidemiological evidence suggesting that vCJD is a new disease, this might be a reasonable assumption to make considering the drawbacks of any retrospective review (lack of medical records, sample size, and limited pathological material). This is not the case. A recent retrospective study12 attempted to examine the extent of misclassification of death from Creutzfeldt-Jakob disease in England in 1485 people aged 15–44 who died from highly selected neurological disorders during 1979–96. The authors concluded that there had not been any, although pathological material was not examined. The advantages of our study are that all ICD9 rubrics broadly compatible with a diagnosis of vCJD were studied from an entire population and pathological material was examined. The study of Majeed et al12 together with this study suggests that the recognition of vCJD in 1996 represents the emergence of a new disease and is not just an artefact of better case ascertainment.
Acknowledgments
We thank Mrs V Robertson, University of Wales College of Medicine; Professor Michel Coleman, London School of Hygiene and Tropical Medicine; Dr AR Freedman, Professor G Lewis, Professor CM Wiles, University of Wales College of Medicine; Dr M Cotter, Health Solutions Wales; consultant pathologists: Dr A W Caslin and Dr D M Wayte, Ysbyty Gwynedd; Dr A D A Dalton, Glan Clwyd Hospital; Dr A Dawson and Dr C J O'Brien, Morriston Hospital; Dr R B Denholm and Dr J K Murphy, West Wales General Hospital; Dr R W Fortt, Royal Gwent Hospital; Dr A R Gibbs, Llandough Hospital; Dr K J Holley, Warwick Hospital; Dr G R Melville-Jones, Withybush General Hospital; Dr L A Murray, Prince Philip Hospital; Dr A Rees, Princess of Wales Hospital; Dr P W B Scott, Nevill Hall Hospital; Dr C G B Simpson, Bronglais General Hospital; Dr D Stock, Royal Glamorgan Hospital; Dr S Williams, Singleton Hospital; Dr A H Burdge and Dr Williams, Wrexham Maelor Hospital; Dr N Johnston and Ms L Conyers, Derriford Hospital; HM Coroners for England and Wales; Medical Directors of NHS Hospital Trusts in Wales; the Directors for Public Health Medicine for Health Authorities in Wales; and all medical records staff who helped in notes retrieval.
Funding was provided by the Department of Health; the views expressed in this paper are those of the authors and are not necessarily those of the Department of Health.
REFERENCES
Linked Articles
- Editorial commentary