Article Text

Abstract
Prions are infectious pathogens principally composed of abnormal forms of a protein encoded in the host genome. They cause lethal neurodegenerative conditions including CJD, GSS, and kuru in humans and scrapie and bovine spongiform encephalopathy in domestic animals. Remarkably, distinct strains of prions occur despite absence of an agent-specific genome: misfolded proteins themselves may encode strain diversity – with wide implications in biology. The arrival of variant CJD, and the experimental confirmation that it is caused by infection with BSE-like prions, has focussed research on early diagnosis and treatment. Recent advances lead to considerable optimism that effective human therapies may now be developed. While several drugs have been tried in small numbers of patients, there is no clear evidence of efficacy of any agent and controlled clinical trials are urgently needed. Importantly, there is increasing recognition that fundamental processes involved in prion propagation – seeded aggregation of misfolded host proteins – are of far wider significance, not least in understanding the commoner neurodegenerative diseases that pose such a major and increasing challenge for healthcare in an ageing population.
- CJD, Creutzfeldt-Jakob disease
- CNS, central nervous system
- FDC, follicular dendritic cell
- GPI, glycosylphosphatidylinositol
- GSS, Gerstmann-Sträussler-Scheinker disease
- LRS, lymphoreticular system
- vCJD, variant CJD
- Creutzfeldt-Jakob disease
- Gerstmann-Sträussler-Scheinker disease
- kuru
- prion
- variant Creutzfeldt-Jakob disease
Statistics from Altmetric.com
- CJD, Creutzfeldt-Jakob disease
- CNS, central nervous system
- FDC, follicular dendritic cell
- GPI, glycosylphosphatidylinositol
- GSS, Gerstmann-Sträussler-Scheinker disease
- LRS, lymphoreticular system
- vCJD, variant CJD
- Creutzfeldt-Jakob disease
- Gerstmann-Sträussler-Scheinker disease
- kuru
- prion
- variant Creutzfeldt-Jakob disease
The prion diseases, or transmissible spongiform encephalopathies, are neurodegenerative conditions that affect both humans and animals. The prototypic disease is scrapie, a naturally occurring disease of sheep and goats, present in many countries worldwide and recognised in Europe for over 200 years. Other animal prion diseases, recognised over the last few decades, include transmissible mink encephalopathy and chronic wasting disease of mule deer and elk (principally in the United States) and, since the 1980s, bovine spongiform encephalopathy (BSE), first described in the UK and now recognised in most European Union countries, Japan, Canada, and the United States. The more recently described feline spongiform encephalopathy of domestic cats and spongiform encephalopathies of a number of species of zoo animals1 are now also recognised as prion diseases.
The human prion diseases have been traditionally classified into Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker disease (GSS), and kuru. Although these are rare disorders, affecting about one to two people per million world-wide per annum, remarkable attention has been focused on them in recent years. This is because of the unique biology of the transmissible agent or prion, and also because of fears that the epizootic of BSE could pose a threat to public health through dietary exposure to infected tissues.
Scrapie was demonstrated to be transmissible by inoculation between sheep (and goats) following remarkably prolonged incubation periods in 1936. It was assumed that some type of virus must be the causative agent and Sigurdsson coined the term “slow virus infection” in 1954. There was considerable interest in the 1950s in an epidemic of a neurodegenerative disease, kuru, characterised principally by a progressive cerebellar ataxia, amongst the Fore linguistic group of the Eastern Highlands of Papua New Guinea. Subsequent field work suggested that kuru was transmitted during cannibalistic feasts. In 1959 Hadlow drew attention to the similarities between kuru and scrapie at the neuropathological, clinical, and epidemiological levels leading to the suggestion that these diseases may also be transmissible.2 A landmark in the field was the transmission, by intracerebral inoculation with brain homogenates into chimpanzees, of kuru and then CJD by Gajdusek and colleagues in 1966 and 1968, respectively.3 Transmission of GSS followed in 1981. This work led to the concept of the “transmissible dementias”. The term “Creutzfeldt-Jakob disease (CJD)” was introduced by Spielmeyer in 1922 drawing from the case reports of Creutzfeldt (1920) and Jakob (1921) and was used in subsequent years to describe a range of neurodegenerative conditions, many of which would not meet modern diagnostic criteria for CJD. Interestingly, Jakob suspected that the condition may be transmissible and experimentally inoculated rabbits in an attempt to demonstrate this in the 1920s. This was unsuccessful and we now know that rabbits are unusually resistant to prion infection.
The criterion of transmissibility allowed diagnostic criteria for CJD to be assessed and refined. All the animal and human conditions share common histopathological features. The classical diagnostic triad of spongiform vacuolation (affecting any part of the cerebral grey matter), neuronal loss, and astrocytic proliferation may be accompanied by amyloid plaques.
The nature of the transmissible agent has been a subject of intense and heated debate for many years. The initial assumption that it must be viral was challenged, however, both by the failure to directly demonstrate a virus (or an immunological response) and because the transmissible agent was resistant to treatments which inactivate nucleic acids (such as ultraviolet radiation or treatment with nucleases). These remarkable findings led to suggestions in 1966 by Tikvar Alper and others that the transmissible agent may be devoid of nucleic acid4,5 and led John Griffith to suggest in 1967 that the transmissible agent may in fact be composed entirely of protein.6 In this remarkable letter to Nature, he proposed three hypothetical mechanisms for propagation of such an agent, one of which closely mirrors current thinking; indeed his model also presciently predicted the existence of distinct strains of agent. Needless to say, such a proposal met with great scepticism at the time, in what was the heyday of the “central dogma” of biology: that DNA encodes RNA that in turn encodes protein.
More than a decade later this remarkable proposal was lent biochemical credibility by intensive purification studies allied with laborious rodent bioassay. Progressive enrichment of brain homogenates for infectivity resulted in the isolation of a protease resistant glycoprotein, designated the prion protein (PrP) by Prusiner and co-workers in 1982. This protein was the major constituent of infective fractions and was found to accumulate in affected brains and sometimes to form amyloid deposits. The term prion (from proteinaceous infectious particle) was proposed7 to distinguish the infectious pathogen from viruses or viroids. Prions were defined as “small proteinaceous infectious particles that resist inactivation by procedures which modify nucleic acids”.
The protease resistant PrP extracted from affected brains was of 27–30 kDa and became known as PrP27–30. Determination of some of the amino acid sequence of PrP27–30 enabled production of oligonucleotides that were used to screen cDNA libraries prepared from scrapie infected hamsters. These studies led to the recovery of cognate cDNA clones by Weissmann and colleagues in 1985. While it had been previously assumed that PrP would be virally encoded, remarkably, PrP27–30 was encoded by a single copy host chromosomal gene rather than by a putative viral nucleic acid in fractions enriched for scrapie infectivity. It then became clear that PrP27–30 was derived from a larger molecule of 33–35 kDa designated PrPSc (denoting the Scrapie isoform of the protein).8 The normal product of the PrP gene however is protease sensitive and was designated PrPC (denoting the Cellular isoform of the protein). No differences in amino acid sequence between PrPSc and PrPC or indeed any consistent covalent differences have been identified and it is clear that PrPSc is derived from PrPC by a post-translational process.9,10
Clinical neurology and neurogenetics have played a major role in the evolution of our understanding of the pathobiology of prion disease. Study of the various forms of human prion disease has been crucial, notably the recognition that the familial forms of the human diseases – already known to be transmissible to laboratory animals by inoculation – are in fact autosomal dominant inherited conditions associated with coding mutations in the gene encoding PrPC (designated PRNP).11,12 The very strong genetic linkage and high or complete penetrance of some such mutations13,14 left no need to propose any external infectious agent and argued strongly for what had become known as the “protein-only” hypothesis of prion propagation.
MOLECULAR BIOLOGY OF PRIONS
Nature of the infectious agent
A number of converging lines of experimental evidence now firmly support the idea that prions consist principally, or entirely, of abnormal isoforms of host encoded prion protein (for a review, see Prusiner15). PrPSc is derived from PrPC by a post-translational mechanism9 and no covalent differences10 between PrPC and PrPSc have been demonstrated. It is proposed that PrPSc acts as a template which promotes the conversion of PrPC to PrPSc and that the difference between these isoforms lies in their conformation and state of aggregation.
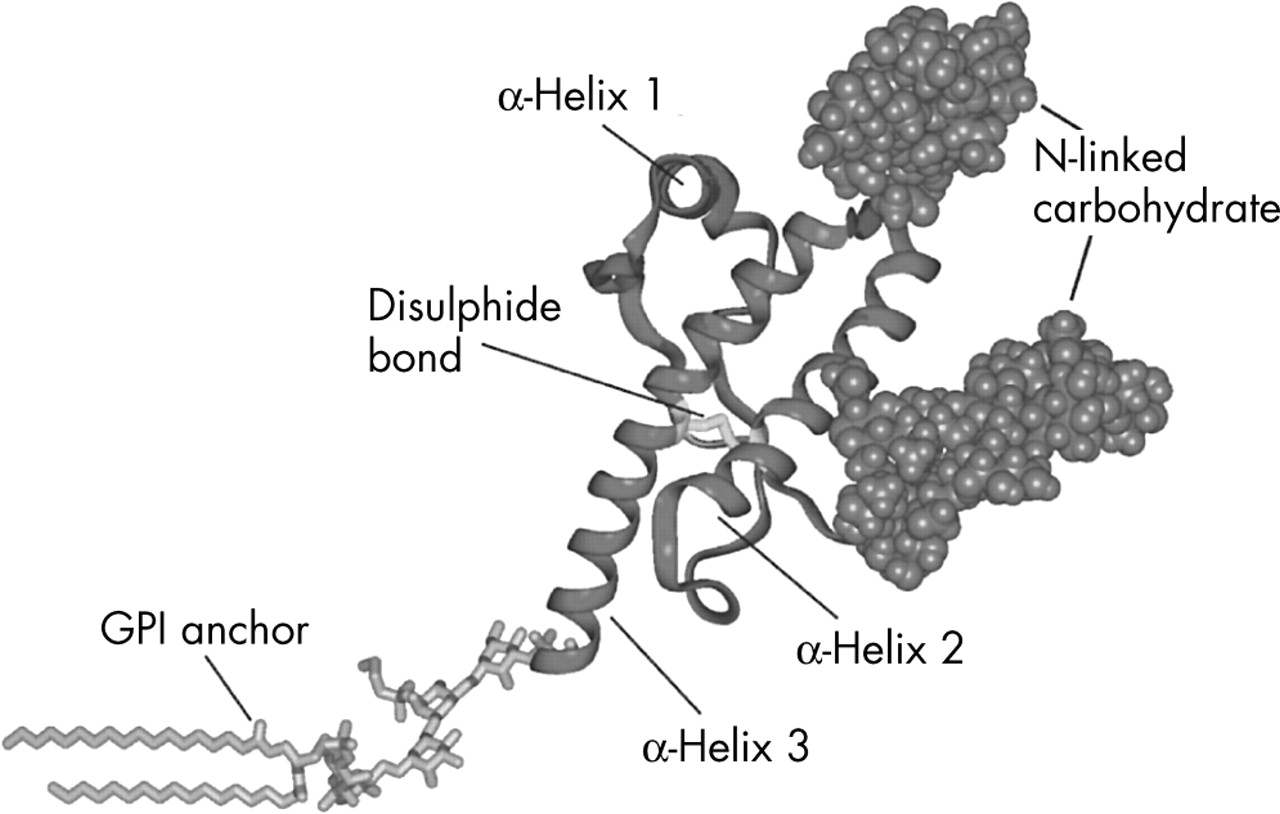
The three dimensional fold or conformation of the cellular isoform was first established by NMR spectroscopy of recombinant mouse PrP.16 Subsequent studies on hamster, human, and other mammalian PrPs show that they have essentially the same conformation. Following cleavage of signal sequences, the mature PrPC consists of an N-terminal region of about 100 amino acids (which is unstructured in the isolated molecule in solution) and a C-terminal domain, also of around 100 amino acids. The C-terminal domain is composed largely of α-helical structure (with three α-helices and a short anti-parallel β-sheet) stabilised by a single disulphide bond linking helices 2 and 3. PrP is a glycoprotein with two asparagine linked glycosylation sites and is attached to the external cell surface via a glycosylphosphatidylinositol (GPI) anchor (fig 1).
The N-terminal region contains five repeats of an 8 amino acid sequence (the octapeptide repeat region). Mutations in this region, resulting in addition of integral numbers of additional repeats, lead to forms of inherited prion disease. While unstructured in the isolated molecule, this region is very highly conserved in evolution and contains two tight binding sites for Cu2+ ions.17 It is proposed that the unstructured N-terminal region may acquire structure following copper binding and a role for PrP in copper metabolism or transport is possible. Disturbance of this function by the conformational transitions between isoforms of PrP could be involved in prion related neurotoxicity.
PrPSc is extracted from affected brains as highly aggregated, detergent insoluble, material that is not amenable to high resolution structural techniques. However, Fourier transform infrared spectroscopic methods show that PrPSc, in sharp contrast to PrPC, has a high β-sheet content.18 The underlying molecular events during infection which lead to the conversion of PrPC to the scrapie agent remain ill defined. The most coherent and general model thus far proposed is that the protein, PrP, fluctuates between a dominant native state, PrPC, and a series of minor conformations, one or a set of which can self associate in an ordered manner to produce a stable supra-molecular structure, PrPSc, composed of misfolded PrP monomers. Once a stable “seed” structure is formed, PrP can then be recruited, leading to an explosive, auto-catalytic formation of PrPSc.
Little is known for certain about the molecular state of the protein that constitutes the self propagating, infectious particle itself. There are examples of infectivity in the absence of detectable PrPSc,19–22 and different strains of prions (see below) are known to differ in their degree of protease resistance. A single infectious unit corresponds to around 105 PrP molecules.23 It is unclear whether this indicates that a large aggregate is necessary for infectivity, or at the other extreme, whether only a single one of these PrPSc molecules is actually infectious. This relationship of PrPSc molecules to infectivity could simply, however, relate to the rapid clearance of prions from the brain known to occur following their intracerebral inoculation.
The difficulty in performing structural studies on native PrPSc has led to attempts to produce soluble β-sheet rich forms of PrP, which may be amenable to NMR or crystallographic structure determination. It is now recognised that the adage regarding protein folding: “one sequence, one conformation”, is not strictly true. Depending on solvent conditions, probably any protein chain can adopt a variety of conformations in which there is a degree of periodic order (that is, extensive regions of secondary structure). However such alternative states do not have precisely and tightly packed side chains which are the hallmark of the native state of orthodox globular proteins.
Studies on a large fragment of the human prion protein (PrP91–231) have shown that at acidic pH PrP can fold to a soluble monomer comprised almost entirely of β-sheet in the absence of denaturants.24 Reduction of the native disulphide bond was a prerequisite for β-sheet formation and these observations of alternative folding pathways dependent upon solvent pH and redox potential could have important implications for the mechanism of conversion to PrPSc. Indeed, this monomeric β-sheet state was prone to aggregation into fibrils with partial resistance to proteinase K digestion, characteristic markers of PrPSc. Unusually for a protein with a predominantly helical fold, the majority of residues in PrP91–231 have a preference for β-conformation. In view of this property, it is possible that the PrP molecule is delicately balanced between radically different folds with a high energy barrier between them; one dictated by local structural propensity (the β-conformation) and one requiring the precise docking of side chains (the native α-conformation). Such a balance would be influenced by mutations causing inherited human prion diseases.
The precise sub-cellular localisation of PrPSc propagation remains controversial. However, there is considerable evidence implicating either late-endosome-like organelles or lysosomes.25–28 The environments of these organelles are evolved to facilitate protein unfolding at low pH prior to degradation by acid activated proteases. It is possible that the α-PrP to β-PrP conversion, caused by reduction and mild acidification, is relevant to the conditions that PrPC would encounter within the cell, following its internalisation during re-cycling. Such a mechanism could underlie prion propagation, and account for the transmitted, sporadic and inherited aetiologies of prion disease (see table 1 and fig 2). Initiation of a pathogenic self propagating conversion reaction, with accumulation of aggregated β-PrP, may be induced by exposure to a “seed” of aggregated β-PrP following prion inoculation, or as a rare stochastic conformational change, or as an inevitable consequence of expression of a pathogenic PrPC mutant that is predisposed to form β-PrP. It remains to be demonstrated whether such alternative conformational states of the protein are sufficient to cause prion disease in an experimental host or whether other cellular co-factors are also required.
Prion strains
In common with other pathogens, distinct naturally occurring isolates or strains are observed. However, it has been unclear how such strain properties could be encoded within an agent devoid of nucleic acids. Dickinson and colleagues isolated multiple distinct strains of naturally occurring sheep scrapie in mice. Such strains are distinguished by their biological properties: they produce distinct incubation periods and patterns of neuropathology in inbred lines of laboratory mice (for a review, see Bruce et al29). As they can be serially propagated in inbred mice with the same Prnp genotype, they cannot be encoded by differences in PrP primary structure. Furthermore, strains can be re-isolated in mice after passage in intermediate species with different PrP primary structures.30 Conventionally, distinct strains of conventional pathogen are explained by differences in their nucleic acid genome. However, in the absence of such a scrapie genome, alternative possibilities must be considered. The protein-only hypothesis,6 if correct, must be able to explain how a single polypeptide chain could encode multiple disease phenotypes. Clearly, understanding how a protein-only infectious agent could encode such phenotypic information is of wide biological interest.
Support for the idea that strain specificity may be encoded by PrP itself was provided by study of two distinct strains of transmissible mink encephalopathy prions which can be serially propagated in hamsters, designated hyper (HY) and drowsy (DY). These strains can be distinguished by differing physiochemical properties of the accumulated PrPSc in the brains of affected hamsters.31 Following limited proteolysis, strain specific migration patterns of PrPSc on polyacrylamide gels were seen which related to different N-terminal ends of HY and DY PrPSc following protease treatment and implying differing conformations of HY and DY PrPSc.32
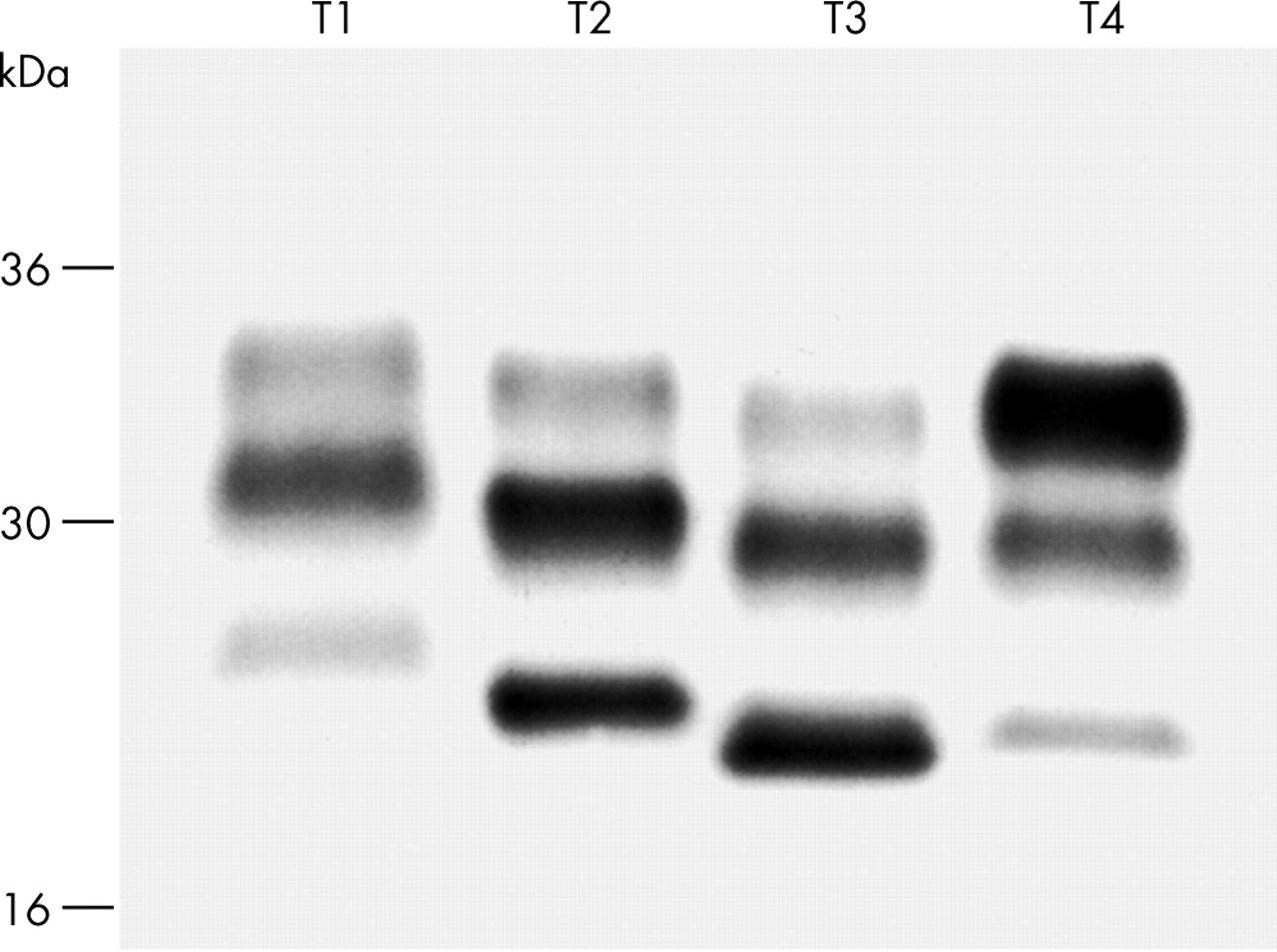
Distinct human PrPSc types have been identified which are associated with different phenotypes of CJD33,34 (fig 3). The different fragment sizes seen on western blots following treatment with proteinase K suggests that there are several different human PrPSc conformations. However, while such biochemical modifications of PrP are clearly candidates for the molecular substrate of prion strain diversity, it is necessary to be able to demonstrate that these properties fulfil the biological properties of strains, in particular that they are transmissible to the PrP in a host of both the same and different species. This has been demonstrated in studies with CJD isolates, with both PrPSc fragment sizes and the ratios of the three PrP glycoforms (diglycosylated, monoglycosylated, and unglycosylated PrP) maintained on passage in transgenic mice expressing human PrP.34 Furthermore, transmission of human prions and bovine prions to wild type mice results in murine PrPSc with fragment sizes and glycoform ratios which correspond to the original inoculum.34 Variant CJD (vCJD) is associated with PrPSc glycoform ratios which are distinct from those seen in classical CJD. Similar ratios are seen in cattle BSE and BSE when transmitted to several other species.34 These data strongly support the “protein-only” hypothesis of infectivity and suggest that strain variation could be encoded by a combination of PrP conformation and glycosylation. Furthermore, polymorphism in PrP sequence can influence the generation of particular PrPSc conformers.34 As PrP glycosylation occurs before conversion to PrPSc, the different glycoform ratios may represent selection of particular PrPC glycoforms by PrPSc of different conformations. According to such a hypothesis, PrP conformation would be the primary determinant of strain type with glycosylation being involved as a secondary process. However, since it is known that different cell types may glycosylate proteins differently, PrPSc glycosylation patterns may provide a substrate for the neuropathological targeting that distinguishes different prion strains.34 Particular PrPSc glycoforms may replicate most favourably in neuronal populations with a similar PrP glycoform expressed on the cell surface. Such targeting could also explain the different incubation periods which also discriminate strains, targeting of more critical brain regions, or regions with higher levels of PrP expression, producing shorter incubation periods. Recent work has shown strain-specific protein conformation can be influenced by the binding of copper and zinc to PrPSc providing a novel mechanism for post-translational modification of PrP, and for the generation of multiple prion strains.35
Molecular strain typing of prion isolates now allows molecular diagnosis of vCJD34,36 and a new classification of human prion diseases with implications for epidemiological studies investigating the aetiology of sporadic CJD (fig 3). Such methods allow strain typing to be performed in days rather than the 1–2 years required for classical biological strain typing. This technique may also be applicable to determining whether BSE has transmitted to other species,34 thereby posing a threat to human health, for instance to sheep.37–39
Such ability of a single polypeptide chain to encode information specifying distinct phenotypes of disease raises intriguing evolutionary questions. Do other proteins behave in this way? The novel pathogenic mechanisms involved in prion propagation may be of far wider significance and relevant to other neurological and non-neurological illnesses. Indeed, other prion-like mechanisms have now been described and the field of yeast and fungal prions has emerged.40,41
The “species barrier”
Transmission of prion diseases between different mammalian species is restricted by a “species barrier”.42 On primary passage of prions from species A to species B, typically not all inoculated animals of species B develop disease, and those that do have much longer and more variable incubation periods than is seen with transmission of prions within the same species, where typically all inoculated animals succumb with a relatively short, and remarkably consistent, incubation period. On second passage of infectivity to further animals of species B, transmission parameters resemble within species transmissions, with most, if not all, animals developing the disease with short and consistent incubation periods. Species barriers can therefore be quantitated by measuring the fall in mean incubation period on primary and second passage, or, perhaps more rigorously, by a comparative titration study. The latter involves inoculating serial dilutions of an inoculum in both the donor and host species and comparing the doses required to kill 50% of inoculated animals (LD50). The effect of a very substantial species barrier is that few, if any, animals succumb to disease at all on primary passage, and then at incubation periods approaching the natural lifespan of the species concerned.
Early studies of the molecular basis of the species barrier argued that it principally resided in differences in PrP primary structure between the species from which the inoculum was derived and the inoculated host. Transgenic mice expressing hamster PrP were, unlike wild type mice, highly susceptible to infection with Sc237 hamster prions.43 That most sporadic and acquired CJD occurred in individuals homozygous at PRNP polymorphic codon 129 supported the view that prion propagation proceeded most efficiently when the interacting PrPSc and PrPC were of identical primary structure.44,45 However, it has been long recognised that prion strain type affects ease of transmission to another species. Interestingly, with BSE prions the strain component to the barrier seems to predominate, with BSE not only transmitting efficiently to a range of species, but maintaining its transmission characteristics even when passaged through an intermediate species with a distinct PrP gene.30 For instance, transmission of CJD prions to conventional mice is difficult, with few if any inoculated mice succumbing after prolonged incubation periods, consistent with a substantial species barrier.36,46 In sharp contrast, transgenic mice expressing only human PrP are highly susceptible to CJD prions, with 100% attack rate and consistent short incubation periods which are unaltered by second passage, consistent with a complete lack of species barrier.46 However, vCJD prions (again comprising human PrP of identical primary structure) transmit much more readily to wild type mice than do classical CJD prions, while transmission to transgenic mice is relatively less efficient than with classical CJD.36 The term “species barrier” does not seem appropriate to describe such effects and “transmission barrier” may be preferable.47 Both PrP amino acid sequence and strain type affect the 3D structure of glycosylated PrP, which will presumably, in turn, affect the efficiency of the protein-protein interactions thought to determine prion propagation.
Mammalian PrP genes are highly conserved. Presumably only a restricted number of different PrPSc conformations (that are highly stable and can therefore be serially propagated) will be permissible thermodynamically and will constitute the range of prion strains seen. PrP glycosylation may be important in stabilising particular PrPSc conformations. While a significant number of different such PrPSc conformations may be possible amongst the range of mammalian PrPs, only a sub-set of these would be allowable for a given single mammalian PrP. Substantial overlap between the favoured conformations for PrPSc derived from species A and species B might therefore result in relatively easy transmission of prion diseases between these two species, while two species with no preferred PrPSc conformations in common would have a large barrier to transmission (and indeed transmission would necessitate a change of strain type). According to such a conformational selection model47 of a prion transmission barrier, BSE may represent a thermodynamically highly favoured PrPSc conformation that is permissive for PrP expressed in a wide range of different species, accounting for the remarkable promiscuity of this strain in mammals. Contributions of other components to the species barrier are possible and may involve interacting co-factors which mediate the efficiency of prion propagation, although no such factors have yet been identified.
Recent data has further challenged our understanding of transmission barriers.48 The assessment of species barriers has relied on the development of a clinical disease in inoculated animals. However, it is now clear that subclinical prion infections are sometimes established on prion inoculation of a second species.49 Such animals may harbour high levels of prion infectivity but do not develop clinical signs of disease during a normal lifespan. The barrier to primary passage may then be to the development of rapid neurodegeneration and the resulting clinical syndrome rather than a barrier to prion propagation itself. The existence of such subclinical carrier states of prion infection has important potential animal and public health implications and argues against direct neurotoxicity of prions.49
Pathogenesis
In some experimental rodent scrapie models, as in natural sheep scrapie, infectivity is first detectable in the spleen and other lymphoreticular tissues (for a review, see Fraser et al50). Spleen titres rise to a plateau early in the incubation period, long before neuroinvasion is detectable. Central nervous system (CNS) prion replication then rises to high levels and the clinical phase follows next. The route of entry of prions following oral exposure may follow invasion of Peyer’s patches and other gut lymphoid tissues; the relative protease resistance of prions presumably allows a significant proportion of infectivity to survive the digestive tract. It is unclear how prions transit the intestinal mucosa, although M cells may be involved.51 It has been suggested that myeloid dendritic cells mediate transport within the lymphoreticular system (LRS).52 While mature B cells are required for peripheral prion propagation, this appears to be because they are required for maturation of follicular dendritic cells (FDCs). PrPSc accumulates in FDCs which are a long lived cell type and it is thought that they are the site of prion propagation in the spleen.53–55 However, neuroinvasion is possible without FDCs, indicating that other peripheral cell types can replicate prions.56,57 Neuroinvasion involves the autonomic nervous system innervating lymphoid tissue with retrograde spread to the spinal cord or via the vagus to the brain stem.58,59 Prions have been detected in the blood at low levels in some rodent models and experimental BSE infected primates60–62 and transmission of BSE prions between sheep by transfusion has been reported.63 Several reports of infrequent transmission from human blood to rodents have been reported (for a review, see Brown64) and there is now evidence for transmission of vCJD prion infection by blood transfusion.65,66
While prominent lymphoreticular involvement is seen in some experimental models or natural prion diseases, it is undetectable in others (for a review, see Fraser et al50). Both host and prion strain effects are relevant. For example, infection of sheep with BSE prions results in a wide tissue distribution of infectivity, while infection of cattle with this strain does not, infectivity being largely confined to the CNS. In humans infected with sporadic CJD prions, infectivity is largely confined to the CNS, while in vCJD there is prominent involvement of lymphoreticular tissues.67–69
APPLICATION TO CLINICAL NEUROLOGY
Molecular classification of prion disease
While neurologists have until recent years had to rely largely on clinical features to differentiate neurodegenerative disorders, the major advances in molecular genetics and in understanding molecular pathogenesis increasingly enable diagnosis using criteria higher in the diagnostic hierarchy of pathology. While prion diseases have traditionally been classified into CJD, GSS, and kuru, we can now divide them aetiologically into inherited, sporadic, and acquired forms (table 1) with sub-classification according to molecular criteria.
The terms CJD, GSS, and kuru simply represent clinical and/or pathological syndromes within a wider spectrum of disease and are also becoming problematic. For example the term CJD, now widely associated in the public mind with BSE, may be misleading. vCJD and familial CJD may be clinically very different to classical CJD and to each other, and of course have radically different aetiologies, both from each other and from classical sporadic CJD.
Around 15% of recognised prion disease is an inherited Mendelian disorder associated with one of the more than 30 recognised coding mutations in PRNP (for a review, see Collinge66). For a single gene inherited disorder of high penetrance such as inherited prion disease, the diagnostic supremacy of direct demonstration of causative mutation by DNA analysis is clear. Indeed, the availability of such definitive diagnostic markers has long allowed diagnosis of inherited prion disease in patients not only atypical on clinical grounds, but in whom classical neuropathological features are absent.71,72 Indeed, the inherited prion diseases were the first neurodegenerative conditions for which direct gene tests were available and definitive pre-symptomatic testing was performed.71,73 Such definite molecular genetic diagnosis has allowed recognition of a wider phenotypic range than hitherto appreciated.74 Kindreds with inherited prion disease have been described with phenotypes of classical CJD, GSS, and also with other neurodegenerative syndromes including fatal familial insomnia.75 GSS is a pathological term referring to the presence of particular amyloid plaque morphologies now recognised to a variable extent in several different inherited prion diseases. Kindreds are documented in which some individuals have the classical syndromes of CJD and GSS, while others do not fit these rubrics at all.13 Cases diagnosed by PrP gene analysis have been reported which are not only clinically atypical but which lack the classical histological features entirely.72 Neuropathology in such patients is no longer the gold standard: rather the recognised clinicopathological manifestation of a particular inherited condition simply widens. Significant clinical overlap exists with familial Alzheimer’s disease, Pick’s disease, frontal lobe degeneration of non-Alzheimer type, and amyotrophic lateral sclerosis with dementia. All the inherited forms can now be definitively diagnosed and sub-classified according to pathogenic mutation.
Sporadic CJD makes up around 85% of all recognised human prion disease. It occurs in all countries with an apparently random distribution and annual incidence of 1–2 per million; hypothesised causes include spontaneous production of PrPSc via rare stochastic events, somatic mutation of PRNP, or unidentified environmental prion exposure. An association with sheep scrapie is not supported by epidemiological studies,76 however the lack of such evidence does not exclude the possibility that a fraction of sporadic CJD is caused by environmental exposure to animal or human prions. There is marked genetic susceptibility in sporadic CJD in that most cases occur in homozygotes at codon 129 of PRNP, where either methionine (M) or valine (V) may be encoded. Heterozygotes (MV) appear significantly protected against developing sporadic CJD.77–79 Additionally, a PRNP susceptibility haplotype has been identified indicating additional, as yet uncharacterised, genetic susceptibility to sporadic CJD at or near to the PRNP locus.80
The acquired prion diseases include iatrogenic CJD and kuru, and arise from accidental exposure to human prions through medical or surgical procedures or participation in cannibalistic feasts. The two most frequent causes of iatrogenic CJD occurring through medical procedures have arisen as a result of implantation of dura mater grafts and treatment with human growth hormone derived from the pituitary glands of human cadavers.81,82 Other iatrogenic cases have resulted from corneal transplantation and use of contaminated neurosurgical instruments.81,82PRNP codon 129 genotype is also relevant to susceptibility and incubation period.77,83 The occurrence of cases of apparently sporadic CJD in unusually young people in the UK in 199584–86 led to concerns that BSE transmission to humans may have occurred. Arrival of further cases in 1996 led to the recognition of a novel clinicopathological type of human prion disease, now known as vCJD,87 indicating the arrival of a new risk factor for CJD in the UK.88 A link with BSE seemed highly likely on epidemiological grounds and this was strongly supported by experimental data, firstly from molecular strain typing studies34 and later by transmission studies into both transgenic and conventional mice.36,89PRNP mutations are absent in vCJD, and all cases studied to date have been 129 MM90,91 (also unpublished data). That vCJD is caused by the same prion strain as that causing BSE in cattle, raised the possibility that a major epidemic of vCJD will occur in the UK and other countries as a result of dietary or other exposure to BSE prions and also concerns of potential iatrogenic transmission of pre-clinical vCJD via medical and surgical procedures.92 That only PRNP 129 MM individuals are susceptible to BSE infection is questionable, since the other acquired human prion diseases, iatrogenic CJD and kuru, occur in all codon 129 genotypes as the epidemic evolves, with MV having the longest mean incubation periods.14,47,93 Human BSE infection of other PRNP genotypes may simply have a longer latency47 (and may also have a different phenotype36).
The acquired prion diseases, such as vCJD, although not contagious in humans, are infectious diseases. In infectious disease, while again clinical and histopathological features may be key, confirmation of diagnosis, not least in life threatening conditions, is by identification of the infectious pathogen itself or a specific immune response to it. Isolation and strain typing of the pathogen is at the apex of the diagnostic hierarchy. Strain typing in particular may allow the source of an outbreak to be identified and the best available prognostic and therapeutic advice to be provided. While it is essential to balance the potential risks and discomfort involved in an invasive diagnostic test against the improved diagnostic accuracy, it will only be by progressing steadily to greater use of molecular analysis of neurological disease that we will be able to deliver the diagnostic and ultimately therapeutic advances to patients with neurodegenerative diseases that are so desperately needed.
The marked phenotypic heterogeneity observed in human prion diseases has yet to be explained. However, it is likely that a significant proportion of this relates to the propagation of distinct human prion strains. The identification of strain specific PrPSc biochemical properties now allows an aetiology based classification by typing of the infectious agent itself. Four types of human PrPSc have now been reliably identified from patients with sporadic or iatrogenic prion disease using molecular strain typing34–36,94 and a preliminary molecular classification proposed94 (fig 3). Sporadic and iatrogenic CJD are associated with PrPSc types 1–3, while type 4 human PrPSc is uniquely associated with vCJD and is characterised by a fragment size and glycoform ratio that is distinct from PrPSc types 1–3 observed in classical CJD.34–36,94 The M/V polymorphism at codon 129 of PRNP is associated with different PrPSc types. PrPSc types 1 and 4 have so far only been detected in MM individuals and type 3 cases are predominantly associated with at least one V allele (MV or VV), while type 2 is seen in any PRNP codon 129 genotype.34,35,68,90,94 An earlier molecular classification33,95 based on subdivision into only two molecular sub-types by fragment size is also in use, and an international consensus has yet to be reached.
Molecular strain typing has major implications for epidemiological surveillance of sporadic CJD, whose aetiology remains obscure. While spontaneous conversion of PrPC to PrPSc as a rare stochastic event, or somatic mutation of the PrP gene, resulting in expression of a pathogenic PrP mutant, are plausible explanations for sporadic CJD,96 other causes for at least some cases, including environmental exposure to human or animal prions, has not been ruled out by existing epidemiological studies.97 Sub-classification of sporadic CJD based upon PrPSc type immediately allows a more precise molecular classification of human prion disease and re-analysis of epidemiological data using these molecular sub-types may reveal important risk factors obscured when sporadic CJD is analysed as a single entity. For example, it will be important to review the incidence of sporadic CJD associated with PrPSc type 2 and other molecular sub-types in both BSE affected and unaffected countries in the light of recent findings suggesting that BSE prion infection of “humanised” transgenic mice may result in propagation of either type 4 PrPSc or type 2 PrPSc.98 Individuals that propagate type 2 PrPSc as a result of BSE exposure may present with prion disease that would be indistinguishable on clinical, pathological, and molecular criteria from that found in classical CJD.
Epidemiology of vCJD and other possible forms of human BSE infection
Estimates of the mean incubation period of human-to-human prion transmission come from study of growth hormone related iatrogenic CJD and kuru: in both cases estimates are of around 12 years. In kuru, incubation periods can exceed 50 years (Whitfield et al, unpublished observations). The effect of a species barrier is to considerably increase both the mean and range of incubation periods seen, which may approach the usual lifespan of the species concerned.47 For example, the cattle-to-mouse barrier for the BSE strain results typically in a three- to fourfold increase in mean incubation period. Mean incubation periods of human BSE infection of 30 years or more should be considered.47 Furthermore, prion disease in mice follows a well defined course with a highly distinctive and repeatable incubation time for a given prion strain in a defined inbred mouse line. In addition to the PrP gene, several additional genetic loci with a major effect on incubation period have now been mapped.93 It can be anticipated that the human homologues of such loci may play a key role in human susceptibility to prion disease, both following accidental human prion exposure and exposure to the BSE agent. By definition, the patients identified to date with vCJD are those with the shortest incubation periods for BSE. These in turn, given that no unusual history of dietary, occupational, or other exposure to BSE has been identified, would be expected to be predominantly those individuals with short incubation time alleles at these multiple genetic loci in addition to having the 129 MM PRNP genotype. The vCJD cases reported to date may therefore represent a distinct genetic sub-population with unusually short incubation periods to BSE prions. It is possible therefore that recent estimates of the size of the vCJD epidemic based on uniform genetic susceptibility may substantially underestimate the eventual size.99,100 In this context, it will be difficult to accurately predict a human epidemic until such loci are identified and their gene frequencies in the population can be determined.101
Long term transmission studies have been carried out using genetically modified mice to both characterise the distinct prion strains causing human disease and to model human susceptibility to infection with BSE and other prions.74 These mice express human, but not mouse, PrPC. While these transgenic mouse models have been able to faithfully propagate human prion strains34,36,46 and recapitulate the characteristic neuropathology of vCJD,98 there are important caveats in extrapolating from such animal models to human susceptibility. However, these studies have found a much higher infection rate in transgenic mice expressing human PrP M129 than mice expressing human PrP V129 when challenged with either BSE or vCJD prions, and demonstrated that BSE prion infection can produce disease phenotypes resembling sporadic CJD infection of these mice and also novel prion strain phenotypes. Most recently, these studies have argued that the vCJD phenotype may only be expressed in the presence of the M form of human PrP.102 While this would imply that only those humans expressing human PrP M129 may develop vCJD, this does not mean that VV individuals are completely resistant to BSE prion infection, but rather that if infected they would show a different phenotype.102
Early diagnosis of human prion disease
Despite the currently dire prognosis of established prion neurodegeneration, it is important to establish a firm early diagnosis. This is firstly because the differential diagnosis of prion disease includes potentially treatable conditions, for example cerebral vasculitis, and cortical biopsy, with full infection control precautions, may be considered in some patients. Secondly, although a devastating diagnosis, early confirmation removes uncertainty, which is itself distressing, obviates the need for further investigation, and allows a care plan to be established including infection control measures and appropriate counselling of patient and their family. However, the advent of the first therapeutic trials in prion disease (see below) creates a new impetus to accelerate diagnosis. Drugs that retard, or even eliminate, prion infection will not reverse existing neuronal loss and there may be little benefit in arresting prion propagation at the advanced stage of neurodegeneration that diagnosis is often made currently (table 2). Advances in early diagnosis must therefore go hand in hand with therapeutic advances to deliver real benefit to patients in the years ahead. While such effective drugs are not yet available, much research is ongoing in both diagnostics and therapeutics. However, given both the rarity and speed of evolution of prion disease, such research can only effectively progress with a high level of early referral, at a stage where the diagnosis may be very unclear, to research centres. For this reason, a UK-wide early referral system has been established in the UK with the support of the Department of Health and the Medical Research Council.
While a confident clinical diagnosis of vCJD may be possible clinically when allied with a positive pulvinar sign on MRI, this sign is not always present, may not be an early feature, and false positives have been reported.103 Further, while the initial patients with vCJD had a relatively uniform clinical presentation, not only are more atypical presentations now becoming apparent, but larger variability can be anticipated with involvement of additional genotypes in BSE prion infection (see above). It will be important to remain open minded about such phenotypes. The early clinical features of vCJD: depression, anxiety, behavioural change, and sensory disturbances, are highly non-specific. Differentiation from much commoner psychiatric causes requires the arrival of overtly neurological features such as ataxia, chorea, and cognitive decline although pre-existing use of neuroleptics and other psychotropic drugs may initially delay their diagnostic recognition. Current clinically based diagnostic criteria for vCJD, undoubtedly important for disease surveillance, require the evolution of disease over at least 6 months and development of several signs indicative of extensive cerebral damage (http://www.doh.gov.uk/cjd/cjd_stat.htm). However, early diagnosis, before extensive irreversible brain damage has occurred, is crucial to exclude an alternative treatable disorder and brain biopsy may well be considered, particularly in younger patients. A clear tissue diagnosis of vCJD can now be made by tonsil biopsy with detection of characteristic PrP immunostaining and type 4t PrPSc69,104 (fig 4). Tonsillar PrPSc is only detectable in vCJD and not other forms of human prion disease. Early tonsil biopsy, if positive, obviates the need for further investigation. While the diagnostic accuracy provided by a tonsil biopsy has to be balanced against the fact that it is an invasive procedure, early referral for investigation should allow much earlier diagnosis and access to clinical trials before extensive functional loss has occurred.
While sporadic CJD can often, following the exclusion of other causes, be diagnosed with a high degree of confidence on the basis of clinical criteria, atypical forms, which present much greater diagnostic difficulty, are not uncommon.105 Relatively slowly progressive forms can be difficult to differentiate from a rapidly progressive form of Alzheimer’s disease with myoclonus for example, and CSF 14-3-3, a marker of neuronal damage, may be unhelpful or misleading in these situations. The large majority of patients with sporadic CJD will be of the PRNP codon 129 MM genotype. Patients with PRNP MV or VV genotypes are more likely to have an atypical course and negative EEG and this may also represent propagation of different prion strain types (see above). Other CSF markers have been proposed including NSE, S-100b, and Tau, but again none are specific for prion disease. There is a pressing need for a non-invasive diagnostic test, ideally of blood, which may allow screening of donated blood and tissue as well as identification of asymptomatically infected and affected individuals. Low levels of PrPSc have been detected in some peripheral tissues, including muscle, in a minority of patients with sporadic CJD at autopsy.106 Such patients were often of long clinical duration and the diagnostic potential of muscle biopsy in the early clinical phase may be limited. Reports of infectivity of blood from patients with classical CJD are infrequent and have been questioned,64 but there is now evidence that vCJD prions may be transmissible by blood transfusion. Such evidence for human prionaemia suggests that detection of PrPSc in human blood may be possible but is below the detection limit of current methods.69 Much research is ongoing to develop more sensitive methods including promising developments with in vitro PrPSc amplification to increase diagnostic test sensitivity.103 Other genetic markers may allow more precise identification of risk groups and both transcriptome and proteome analysis in sporadic and vCJD may provide disease markers.
Presymptomatic and ante-natal testing
Since a direct gene test has become available, it has been possible to provide an unequivocal diagnosis in patients with inherited forms of the disease. This has also led to the possibility of performing pre-symptomatic testing of unaffected but at risk family members, as well as antenatal testing.73 Because of the effect of PRNP codon 129 genotype on the age of onset of disease associated with some mutations, it is possible to determine within a family whether a carrier of a mutation will have an early or late onset of disease. Most of the mutations appear to be fully penetrant, however experience with some is extremely limited. In families with the E200K mutation and in D178N (fatal familial insomnia), there are examples of elderly unaffected gene carriers who appear to have escaped the disease. Genetic counselling is essential prior to pre-symptomatic testing and follows a protocol similar to that established for Huntington’s disease. A positive PrP gene analysis has important consequences for other family members, and it is preferable to have discussed these issues with others in the immediate family before testing. Following the identification of a mutation, the wider family should be referred for genetic counselling. It is vital to counsel both those testing positive for mutations and those untested but at risk, that they should not be blood or organ donors and should inform surgeons, including dentists, of their risk status prior to significant procedures as precautions may be necessary to minimise risk of iatrogenic transmission.
Prevention and public health management
While prion diseases can be transmitted to experimental animals by inoculation, it is important to appreciate that they are not contagious in humans. Documented case-to-case spread has only occurred by cannibalism (kuru) or following accidental inoculation with prions. Such iatrogenic routes include the use of inadequately sterilised intracerebral electrodes, dura mater and corneal grafting, and from the use of human cadaveric pituitary derived growth hormone or gonadotrophin. As discussed above, there is now evidence that vCJD prion infection is transmissible by blood transfusion. UK policy for some time has been to leucodeplete all whole blood and to source plasma for plasma products from outside the UK.
A further possible route of transmission of vCJD is via contaminated surgical and medical instruments. Prions resist conventional sterilisation methods and neurosurgical instruments are known to be able to act as a vector for prion transmission: several cases of iatrogenic transmission of sporadic CJD prions via neurosurgical instruments are documented.107,108 Recent evidence suggests that classical CJD may also be transmitted by other surgical procedures.97 The wider tissue distribution of prions in vCJD69 together with the potential that significant numbers in the population may be silently infected has considerably increased these concerns. Prions adhere avidly to stainless steel and transmit the disease readily in experimental models.109 Tonsillar PrPSc is readily detectable in all cases of vCJD studied at autopsy and lymphoreticular involvement is a very early feature of natural prion infection in sheep and in experimental scrapie models, where replication in the LRS is detectable early in the incubation period and rises to a plateau, which considerably precedes, and is maintained in, the clinical phase.50 This suggests that tonsillar PrPSc has probably been present for a considerable period, perhaps years, before clinical presentation of vCJD in humans, thereby providing the basis of prevalence screening of the general population for infection. Two anonymous screens of tonsil and appendix tissues, removed during routine surgery, have been performed. Tonsil appears a more sensitive reporter of vCJD prion infection than appendix,110 and it is of concern that three positives were reported from a screen of around 12 000 largely appendix samples.111 A pilot scale study of 2000 tonsils found no positives, despite use of high sensitivity methods.112 A national scale study is now being organised by the UK Department of Health to obtain more accurate estimates of prion infection in the UK population.
Certain occupational groups are at risk of exposure to human prions, for instance neurosurgeons and other operating theatre staff, pathologists and morticians, and histology technicians, as well as an increasing number of laboratory workers. Because of the prolonged incubation periods to prions following administration to sites other than the CNS, which is associated with clinically silent prion replication in the lymphoreticular tissue,113 treatments inhibiting prion replication in lymphoid organs may represent a viable strategy for rational secondary prophylaxis after accidental exposure. A preliminary suggested regimen is a short course of immunosuppression with oral corticosteroids in individuals with significant accidental exposure to human prions.105
Therapeutic strategies
All recognised prion diseases are invariably fatal following a relentlessly progressive course. However, there have been significant recent advances in understanding prion propagation and neurotoxicity and clear proof of principle studies of several therapeutic or secondary prophylactic approaches in animal models suggesting effective therapeutics for human disease is realistic.114
While recent attempts at therapeutics in experimental models have focussed on drugs that target PrPSc formation, the cause of cell death in prion neurodegeneration remains unclear. A key area of controversy is whether PrPSc is itself directly neurotoxic. There are prion diseases in which PrPSc levels in brain are very low and, conversely, subclinical prion infection occurs with high levels of PrPSc and no clinical signs.48,49 Also, PrPSc itself is not directly toxic to neurons that do not express PrPC.115 However, the conversion of PrPC to PrPSc is clearly central to prion pathogenesis as mice devoid of PrP (Prnp0/0) are resistant to prion disease and do not propagate infectivity.116,117 An alternative possibility is that neurodegeneration results, at least in part, from loss of normal PrPC function as it is sequestered into PrPSc aggregates, or alternatively, that a toxic intermediate (designated PrPL) may be formed during prion propagation.49 Although PrP-null (Prnp0/0) mice are essentially normal,118,119 adaptive mechanisms during neurodevelopment might compensate for loss of PrPC function, masking any phenotype, whilst loss of PrPC function in the developed, adult CNS might be deleterious. However, this alternative has been effectively excluded by the demonstration that PrPC depletion in the neurons of adult mice also results in healthy animals with no evidence of neuronal loss.120 Importantly, this model validates PrPC as a therapeutic target in prion disease. Also the availability of this model, in which PrP expression in neurons is lost at around 12 weeks post-natally, allows the effect of neuronal PrP ablation during established CNS prion infection to be studied. Remarkably, such mice were completely protected from the development of clinical disease and early spongiform neuropathology was reversed.11 These animals continued to accumulate PrPSc in non-neuronal glial cells and indeed accumulated prions to reach infectious titres seen in end stage conventional prion disease, again arguing against direct neurotoxicity of non-neuronal PrPSc. It is conceivable that reversal of early spongiosis in human disease may even allow some symptomatic recovery. Clearly, the transgenic approach used in this model to interfere with neuronal PrPC expression is not applicable therapeutically in humans, but provides a clear proof of principle for agents or other methods to target PrPC directly. One approach is the isolation of small molecule PrP ligands which bind and stabilise PrPC,74 rendering it less available for conversion, including antibodies that bind or sequester PrPC, or methods to down regulate PrP transcription or translation.
While the precise molecular events involved in the conversion of PrPC to PrPSc remain ill defined, it can be argued that any ligand which selectively stabilises PrPC should block prion propagation. The use of hydrogen/deuterium exchange to measure the extent to which regions of PrPC transiently unfold argues that the conversion of PrPC to PrPSc must proceed through a highly unfolded state that retains little organised native structure and that compounds which can bind to any ordered region of PrPC should therefore inhibit the conversion pathway.121 The maintenance of effective brain levels of such drugs, to reduce prion propagation rates to below those of natural clearance mechanisms, could then plausibly cure prion infection. High throughput screening of large compound libraries can be applied to detect such ligands. This approach has been exploited therapeutically for at least two proteins where mutations or altered conformation result in disease, the central core of p53 and transthyretin.
Considerable progress has been made in immunotherapeutic approaches. Antibodies against several PrP epitopes inhibit PrPSc propagation in cell culture.122–124 These antibodies had little or no affinity for native PrPSc, and might act by binding cell surface PrPC and reducing its availability for incorporation into propagating prions. Transgenic mice expressing anti-PrP μ chains directed against similar epitopes are protected against peripheral (but not central) prion infection.125 In a clear proof of principle, wild type mice that had been peripherally infected with prions were passively immunised with anti-PrP monoclonal antibodies. PrPSc levels and prion infectivity in the spleens of scrapie infected mice were markedly reduced even when antibodies were administered at the point of near maximal PrPSc accumulation. Furthermore, treated animals remained healthy >300 days after untreated animals had succumbed to the disease.126 Unsurprisingly, as antibodies do not readily cross the blood-brain barrier, there was no protective effect in intracerebrally infected mice. Nevertheless, humanised anti-PrP monoclonal antibodies might be used for post-exposure prophylaxis of particular risk groups. For established clinical disease, such antibodies could in principle be given by intracerebroventricular infusion, although effective tissue penetration throughout the CNS would be a problem. An important caveat is that injection of large quantities of anti-PrP antibodies into the CNS caused massive neuronal apoptosis in one study,127 and it will be important to characterise the epitope and dose dependency of this effect.
Active immunisation is limited by immune tolerance to PrP, which is a self protein widely expressed in the immune system. Approaches to overcome this tolerance are being actively investigated, and studies have reported modest protective effects.128,129
The rarity of human prion diseases and consequent difficulty in assessing safety and efficacy means that the development of immunisation methods for healthy individuals to protect against prion diseases is probably not realistic. However, such a programme might eradicate endemic animal prion diseases such as ovine scrapie. It should also be mentioned that clinical trials of active immunisation for Alzheimer’s disease advise caution: anti-Aβ40−42 antibodies can have protective or noxious effects in cells130 and a recent anti-Ab40–42 vaccination trial was halted owing to encephalitic illness in some recipients.131
Other possible future approaches to target PrPC include the use of small duplex RNA molecules to silence gene expression in a sequence specific manner – RNA interference (RNAi). This is a potentially powerful method for therapeutic gene silencing which has been applied to suppress gene expression both in vivo and in vitro in mammalian cells,132,133 including non-dividing cells such as neurons. The prospect of lentiviral mediated RNA silencing of PrPC as a therapeutic tool is conceivable and provides a potential means of extrinsically manipulating PrP gene expression. However, any successful therapeutic delivery system would require effective CNS penetration.
As discussed already, prion strains show considerable diversity and are associated with distinct PrPSc types, which differ in their conformation32–34,134 and glycosylation.34 Strain switching or “mutation” is well documented29,34,36,135 and more than one identifiable strain may propagate in the same host. BSE prions induce replication of two distinct prion strains in transgenic mice expressing only human PrP.98 The possibility of emergence of drug resistance can therefore be anticipated against agents targeting PrPSc by selection of a minor or sub-strain of prion. However, according to the “protein-only” hypothesis, targeting native PrPC should block replication of all prion strains.
The advent of clinical trials in human prion disease
A variety of drugs have been tried in individual or small numbers of patients over many years. With a notable exception,136 reports have been largely anecdotal. There is no clear evidence of efficacy of any agent, and there is no doubt that controlled clinical trials are urgently needed. Such trials will be highly challenging for several reasons. Prion diseases are rare, often rapidly progressive, and always fatal which may make randomisation to placebo unacceptable. Patterns of disease overall are, however, extremely variable with clinical durations varying from weeks to >2 years in sporadic CJD, and >20 years in some inherited prion diseases.74 As “first generation” treatments proposed for prion disease are likely to have only a modest effect on disease progression, even using survival duration as an outcome measure requires study of large numbers to reliably assess efficacy. There is a lack of systematic natural history studies of disease progression and an absence of biological markers of disease activity. The high profile of these diseases often leads to media reporting of putative prion therapeutics with optimistic headline coverage leading, understandably, to patient requests for immediate access and unrealistic expectations of efficacy, particularly in patients with advanced neurodegeneration. When such promise of dramatic effect is not rapidly realised, loss of interest in participation in controlled trials may then follow, which necessarily take considerable time to be planned, peer reviewed, and ethically approved, to assess if the drug actually has a worthwhile therapeutic effect. In the UK, at the request of the Government’s Chief Medical Officer, a clinical trial protocol (http://www.controlled-trials.com/isrctn/trial/PRION/0/06722585.html) and infrastructure has been developed to rigorously assess the drug quinacrine137 and to provide a framework for assessment of novel therapeutics as these become available – the MRC PRION-1 trial. Importantly under these circumstances, a formal consultation with patients’ representatives was organised to refine the protocol so that it would be acceptable to the majority of potential participants (http://www.mrc.ac.uk/prn/pdf-cjd_workshop.pdf). Pentosan polyphosphate is another candidate anti-prion drug and has shown some efficacy in animal models.138,139 Unlike quinacrine, it dues not enter the CNS readily and has been administered by intraventricular infusion in several patients. Major toxicity has been reported by this route in animal studies and such treatment was not supported by the Committee of Safety on Medicines or CJD Therapy Advisory Group in the UK.140,141
Perspectives for the decade ahead
-
Effective treatment of neurodegenerative disease is one of the major challenges facing biomedical research. The focus on prion diseases following the BSE epidemic, added to the intrinsic interest in their unique pathobiology, has led to these diseases being amongst the best understood causes of neurodegeneration.
-
It is to be hoped prion diseases remain rare. If no epidemic of BSE related human infection emerges, such advances in understanding neurodegenerative processes should have far wider value in commoner conditions, such as Alzheimer’s and Parkinson’s disease, also associated with aggregation of misfolded protein. Conversely, if extensive human infection with BSE prions emerges in exposed populations, and silent secondary transmission is efficient, the failure to have developed a treatment for what is, albeit inadvertently, a man-made disease, during the window of opportunity provided by the remarkable latency of these infections would not enhance public perception of biomedical research.
-
Considerable uncertainty remains over the number of people pre- or sub-clinically infected with BSE prions and what proportion of these will develop clinical disease in the years ahead: the natural evolution of this infection in the UK population is likely to span decades. Identification of the key genes controlling incubation period will allow better estimates.
-
Clinicians should remain alert to alternate phenotypes of infection with BSE prions, which could also manifest as sporadic CJD or novel phenotypes as well as vCJD. Molecular classification of prion disease, by both genetic typing of host and strain typing of infecting prion, is evolving and will allow more precise epidemiological studies.
-
Blood-based diagnostic tests will probably become available, involving surrogate serum markers, high sensitivity detection of PrPSc, or in vitro prion amplification. The arrival of such tests, particularly if associated with a significant false positive rate, will raise important ethical issues about anonymity of testing and notification which should be debated in advance.
-
Effective means to decontaminate prions on surgical and medical instruments will become available over the next few years and should reduce the risk of iatrogenic prion transmission. Secondary transmission of vCJD will continue until a suitable screening test for the infected state arrives.
-
Proof of principle studies in animal models suggest that humanised anti-PrP monoclonal antibodies could be used for passive immunisation in early pathogenesis to block neuroinvasion. This treatment could be considered for known iatrogenically infected individuals. Active human immunisation may be feasible technically but is unlikely to be pursued commercially.
-
While the precise molecular events in prion propagation are not clear and the atomic level structure of prions is not yet determined, it is clear that PrPC is the essential substrate. It is also clear that interference with PrPC expression in adult brain is without serious effect and blocks onset of neurological disease in animal models. It should be possible to identify small molecules which penetrate the CNS to bind to PrPC and to prevent its recruitment into prions, or to use one of a number of emerging technologies to reduce PrPC expression in brain. If such methods are able to reduce prion propagation rates to below those of natural clearance mechanisms, it ought to be possible to cure prion infection. New methods for early diagnosis, and their timely use, will be crucial, as such methods will not reverse neuronal cell loss which is appreciable or severe by the time clinical diagnosis is typically reached.
APPENDIX
Glossary
Codon 129 polymorphism: unique to humans, there are two common forms of PRNP encoding either methionine or valine at codon 129; a major determinant of genetic susceptibility to and phenotypic expression of prion disease
Conformational selection model: a hypothetical model which explains transmission barriers on the basis of overlap of permissible conformations of PrPSc (prion strains) between mammalian species
Molecular strain typing: a means of rapidly differentiating prion strains by biochemical differences in PrPSc
Prion: the infectious agent causing prion diseases
Prion incubation period: the interval between exposure to prions and the development of neurological signs of prion disease; typically months even in rodent models and years to decades in humans
Prion protein (PrP): a glycoprotein encoded by the host genome and expressed in many tissues but especially on the surface of neurons
Prion strain: distinct isolates of prions originally identified and defined by biological characteristics which breed true in inbred mouse lines
Protein-only hypothesis: that prions lack a nucleic acid genome, are composed principally or solely of abnormal isomers of PrP (PrPSc) and replicate by recruitment of host PrPC
PRNP: the human prion protein gene; mouse gene is designated Prnp
PrPC: the normal Cellular isoform of PrP rich in α-helical structure
PrPSc: the “Scrapie” or disease associated isoform of PrP which differs from PrPC in its conformation and is generally found as insoluble aggregated material rich in β-sheet structure
Species barrier: better called the transmission barrier, this describes the observation that transmission of prions from one species to another is generally inefficient when compared to subsequent passage in the same host species
Subclinical infection: a state where host prion propagation is occurring but which does not produce clinical disease during normal lifespan; essentially a carrier state of prion infection
TSE: transmissible spongiform encephalopathy or prion disease
Key points for clinical practice
Classification of human prion disease
Diagnosis of prion disease

Model of the C-terminal domain of human prion protein indicating positions of N-linked glycans, the single disulphide bond joining helices 2 and 3, and the GPI anchor which attaches the protein to the outer surface of the cell membrane.

Possible mechanism for prion propagation. Largely α-helical PrPC proceeds via an unfolded state (i) to re-fold into a largely β-sheet form, β-PrP (ii). β-PrP is prone to aggregation in physiological salt concentrations. Prion replication may require a critical “seed” size. Further recruitment of unfolded PrP (iii) or β-PrP monomers (iv) then occurs as an essentially irreversible process driven thermodynamically by intermolecular interactions.

Molecular strain typing of human prions. Western blot of brain homogenate after treatment with proteinase K shows different apparent molecular mass and glycoform ratios in patients with forms of sporadic or iatrogenic (T1–3) or vCJD (T4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tonsil biopsy for diagnosis of vCJD prion infection. Western blot demonstrates PrPSc after proteinase K (PK) digestion. Characteristic PrPSc type is seen in tonsil from patients with vCJD allowing firm ante mortem tissue diagnosis.
REFERENCES
Footnotes
-
Competing interests: none declared