Article Text

Abstract
This review aims to update the reader on advances in the understanding of haematological conditions that may arise in neurological practice. Thrombophilia, antiphospholipid antibody syndrome, thrombotic thrombocytopenic purpura, sickle cell and clonal disorders associated with neuropathy are discussed.
- aPL, antiphospholipid antibodies
- APS, antiphospholipid syndrome
- FVL, factor V Leiden
- HIT, heparin induced thrombocytopenia
- IL, interleukin
- LA, lupus anticoagulant
- MGUS, monoclonal gammopathy of undetermined significance
- MTHFR, methylene tetrahydrofolate reductase
- PFO, patent foramen ovale
- POEMS, polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes
- PTGM, prothrombin gene mutation
- TTP, thrombotic thrombocytopenic purpura
- VTE, venous thromboembolism
- VWF, Von Willebrand factor
- WM, Waldenströms macroglobulinaemia
Statistics from Altmetric.com
- aPL, antiphospholipid antibodies
- APS, antiphospholipid syndrome
- FVL, factor V Leiden
- HIT, heparin induced thrombocytopenia
- IL, interleukin
- LA, lupus anticoagulant
- MGUS, monoclonal gammopathy of undetermined significance
- MTHFR, methylene tetrahydrofolate reductase
- PFO, patent foramen ovale
- POEMS, polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes
- PTGM, prothrombin gene mutation
- TTP, thrombotic thrombocytopenic purpura
- VTE, venous thromboembolism
- VWF, Von Willebrand factor
- WM, Waldenströms macroglobulinaemia
Although there are numerous neurological manifestations of primary haematological disease this review aims to update the reader on advances in the understanding of haematological conditions that may cross into day to day neurological practice but not to discuss the neurology in detail. We have concentrated on thrombophilia, antiphospholipid antibody syndrome, thrombotic thrombocytopenic purpura, sickle cell and clonal disorders associated with neuropathy. Further information may be obtained from the British Committee for Standards in Haematology (BCSH), which can be accessed online (www.bcshguidelines.com).
HYPERCOAGULABILTY AND THROMBOPHILIA
The process of haemostasis and thrombosis is tightly regulated through the interaction of platelets, coagulation factors and the endothelial lining of blood vessels. Failure of this regulation may result in a hypercoagulable state, characterised by either venous or arterial thrombosis. Arterial thrombosis is associated with platelet reactivity and endothelial wall damage while thrombus formation in the venous system is related to deposition of fibrin and stasis. Hypercoagulability plays much more of a role in venous thrombosis.
Venous thromboembolism
The incidence of venous thromboembolism (VTE) is approximately 117 per 100 0001 person years, and it usually manifests as deep vein thrombosis or pulmonary embolism. VTE occurs because of the combined interaction of environmental and genetic risk factors.2 Environmental factors include high-risk situations (immobilisation, surgery, trauma, travel, oestrogen), predetermined states (advancing age and male sex) and physical states (obesity, malignancy, autoimmune disease and pregnancy). Inherited risk factors increase the likelihood of VTE occurring during high-risk periods, and can also account for the development of thrombosis in the absence of any environmental factors (idiopathic VTE).
A large number of inherited coagulation protein defects are known to influence the tendency to thrombosis. These include deficiencies of the natural anticoagulants (protein C, protein S and antithrombin) which, although uncommon (prevalence of 0.02–0.2%), cause a strong thrombophilic tendency. Procoagulant “gain of function” mutations are less thrombophilic than the anticoagulant deficiencies but are more common and therefore account for a substantial proportion of VTE. These are factor V Leiden (FVL; a mutant factor V (1691 G-A) that is more resistant to inactivation by activated protein C) and the G20210A prothrombin gene mutation (PTGM) which results in elevated prothrombin levels. The relative risks of first VTE for these inherited thrombophilias are outlined in table 1, as published by the retrospective Leiden Thrombophilia Study3 and an American prospective cohort study of over 20 000 subjects.4,5
High concentrations of factor VIII, other haemostatic factors (Von Willebrand factor (VWF), and factors V, VII, FIX and FXI) and D-dimer (a marker of cross linked fibrin breakdown) are also associated with a history of thrombosis, and often follow a hereditary predisposition. Acquired factors can also influence the level of coagulation proteins (liver disease, vitamin K deficiency). Finally, homocysteine, at high concentrations, is also associated with venous thrombosis via genetic mechanisms (C677T 5, 10 methylene tetrahydrofolate reductase (MTHFR) or cystathione β-synthetase mutations) and/or acquired factors (deficiencies of folic acid, or vitamin B6 or B12). A detailed review of inherited thrombotic disorders can be found elsewhere.6
Following a first VTE event, subsequent events can occur in patients with or without a documented thrombophilic condition, with the highest risk in the first 6 months. A cumulative incidence of VTE as high as 25% after five years of follow up is frequently quoted7 although the Leiden Thrombophilia Study found a cumulative incidence of 12.4% after 5 years.8 Despite the risk of an inherited thrombophilia and first VTE being well known, the relative risk for recurrent VTE has until recently remained unclear. This is important to establish as physicians must attempt to accurately predict the risk of further thrombosis in order to make appropriate decisions regarding treatment. High quality studies9,10 have failed to demonstrate a strong influence of inherited prothrombotic states on recurrence of VTE in unselected patients. The Leiden Study Group concluded that prothrombotic abnormalities do not appear to play an important role in the risk of recurrence, and that factors including sex, oral contraceptive use, family history, extent of residual thrombosis, idiopathic versus provoked thrombosis, and D-dimer levels may be far more important. Therefore, the use of thrombophilia testing has become increasingly questioned with regard to its usefulness in predicting subsequent risk of VTE.
There remains a limited role for thrombophilia testing (table 2). Interpretation of results must be in concert with the provision of clear and detailed advice based on an appreciation of the limitations of the laboratory tests. Testing can provide patients and their relatives with improved understanding of their disease and the measures needed to avoid further thrombotic episodes. It can assist in decisions regarding future optimal thromboprophylaxis during other periods of increased risk (in particular, oestrogen therapy and pregnancy). It can identify patients with combined defects who may benefit from long term anticoagulation. Testing has limited value in the acute setting as it is unlikely to alter initial management, and the levels of a number of factors will be affected by the recent thrombosis and anticoagulant therapy. Testing can be performed at least 1 month after stopping anticoagulant treatment. Thrombophilia assessment should be considered in all idiopathic first events, recurrent events, those with a family history of VTE, thrombosis in the young and in patients with thrombosis at an unusual site. Given that there is little clinical trial evidence to support specific recommendations for long term treatments of idiopathic VTE based on the results of thrombophilia testing, decisions regarding duration of anticoagulation remain complex and range from 3 months of warfarin following provoked proximal deep vein thrombosis to long term, based on individual clinical factors.11
Tests for the assessment of thrombophilia
The same schema can be applied to cerebral venous thrombosis of which 20% remains idiopathic. The accepted treatment of intracranial venous thrombosis is therapeutic anticoagulation, whether or not haemorrhage is present. The evidence for this is controversial. Early studies suggested that patients treated with anticoagulants had a 15% mortality compared with 69% for those untreated.12 A small prospective trial and retrospective analysis also suggested a significant survival benefit with anticoagulation, and also the safety of heparin in the presence of haemorrhage. Later studies have suggested no advantage of heparin but were weighted toward less severely affected patients with indolent presentations.13
Arterial thrombosis
There is no conclusive evidence that inherited thrombophilias are associated with arterial thromboembolism. Although both FVL and PTGM may be more prevalent among paediatric stroke patients compared with control subjects, prospective studies are lacking.14 A number of case control studies did not find an association between FVL and stroke/transient ischaemic attacks (regardless of age)15 while others suggested either a non-significant trend or an association with the younger age group and stroke or other highly selected populations. Similarly, a case control study investigating PTGM and stroke suggested that it was more common in the younger stroke patient compared with controls.16 The data are conflicting but the common trend is that the relative effect of any thrombophilic state can be seen easily in the young who lack the strong conventional risk factors of stroke. Although not arterial per se, the only strong association between cerebral events and FVL or PTGM is seen in cerebral venous thrombosis. In studies involving myocardial infarction, the PTGM and FVL may be more prevalent among patients with myocardial infarction who do not have atherosclerosis or other conventional risk factors compared with controls, but again there is a lack of compelling evidence and no prospective trials. With regard to deficiencies of the natural anticoagulants (protein C, protein S and antithrombin), largely because of their low prevalence, their role in ischaemic stroke and myocardial infarction is poorly defined and limited to anecdotal reports. Isolated cases of stroke in patients with dysfibrinogenaemia or plasminogen deficiency have also been reported. Although not evidenced based, there is a limited role for evaluating inherited thrombophilic states in stroke patients and the issue of paradoxical embolus is discussed later.
The most solid evidence for hypercoagulability and arterial thrombosis exists for aberrations in homocysteine. Based on over 20 case control studies, homocysteine elevation is associated with a 2–3-fold increased risk of ischaemic stroke.17 Typically, the high homocysteine levels are caused by either a genetic variation of enzymes involved in homocysteine metabolism (eg, C677T MTHFR) or deficiencies of vitamin cofactors (B6, B12 and folate). The most severe manifestations of elevated homocysteine occur in congenital homocystinuria in which patients present with premature atherosclerosis and thromboembolism. Homozygosity for the C677T MTHFR mutation, in the presence of folate deficiency, may lead to hyperhomocysteinaemia but interestingly, studies do not support an increased risk of stroke in association with the C677T MTHFR mutation.18
Paradoxical embolus
The association between thrombophilia and ischaemic stroke remains controversial even in the context of the putative mechanism of a paradoxical embolism. Hypothetically, coexistent patent foramen ovale (PFO) (or another right to left circulatory communication) and deep vein thrombosis could potentiate the risk of stroke. Certainly, mixed opinion exists as to whether there is a higher rate of thrombophilic defects in patients with PFO and ischaemic stroke. Some groups suggest that most inherited prothrombotic states are associated with stroke and PFO while prospective cohort studies have failed to find an association. Karttunen et al compared 58 cryptogenic stroke patients with PFO with 104 matched control patients in Finland and concluded that PTGM and FVL were associated with stroke (p = 0.022) in this setting.19 Others suggest that PTGM is the only thrombophilic defect shown to coexist with PFO in stroke patients.20 Given that less than 10% of all peripheral vein thromboses are identified radiologically, associating deep vein thrombosis with cryptogenic stroke patients and PFO is likely to be difficult. Cramer et al recently found that the incidence of pelvic deep vein thrombosis was significantly higher in patients with cryptogenic stroke (20%), and cryptogenic stroke and PFO (22%), than in patients with stroke of determined causes (4%).21 The mechanism of stroke associated with PFO or atrial septal aneurysm is unclear22 and there remains a lack of convincing evidence to prove or refute the theory of paradoxical embolus.
ANTIPHOSPHOLIPID SYNDROME
The antiphospholipid syndrome (APS) is an acquired thrombophilic autoimmune condition characterised by the presence of antiphospholipid antibodies (aPL), a group of heterogenous autoantibodies directed mainly against phospholipid binding proteins, and associated plasma proteins. The syndrome is defined as the association of these antibodies with the development of thrombosis (arterial or venous) and/or adverse obstetric events. aPL can be found in association with infection (usually transient and non-pathogenic) and autoimmune disease (particularly prevalent in SLE) where the term secondary APS has been coined.
Venous thrombosis is the most common clinical manifestation of APS, and up to half of these patients have pulmonary emboli. Arterial thrombosis presents as ischaemia or infarction with the brain the most common organ involved. Coronary events account for an additional 25% of arterial events and the remainder involve a range of arterial beds (subclavian, renal, retinal, pedal). The frequency of cardiac valvular abnormalities in APS seems to be high.23
The neurological associations of APS/aPL are diverse24,25 and include arterial ischaemic stroke, cerebral venous thrombosis, and neuropsychiatric and movement disorders. Substantive evidence for many reported associations is lacking and the finding of aPL per se needs to be interpreted with caution. For example, in the general stroke population the presence of antibodies does not raise the risk of recurrence over controls.26 Whereas patients with APS constitute a clear group, as do those with coincidental antiphospholipid antibodies, there is a grey area in between, and in some patients the clinical significance of antibodies is conjectural.
There are currently three aPL tests that may be clinically useful for determining the likelihood of thrombosis and assisting in management decisions (lupus anticoagulant (LA) detection, anticardiolipin and anti-β2GP-1 ELISAs). Meta-analyses suggest that LA is the strongest predictor of thrombosis (equal risk of venous or arterial), followed by anti-β2GP1 antibodies (correlates more with arterial thrombosis).27 Anticardiolipin antibodies are the weakest predictor of thrombosis, and correlate better with venous than arterial thrombosis. Some patients may have positive results for all three tests while others have a single isolated positive aPL test, with anticardiolipin antibodies present in approximately 25% of patients with APS who are negative for LA and anti-β2GP-1.28 Therefore, it is important to perform all three tests when suspicious of APS. It is suggested that the presence of multiple aPL is associated with a higher risk of thrombosis than is a solitary antibody.
Anticoagulation is central to the management of APS but primary prevention in individuals with persistent aPL is not supported by the available evidence. Aspirin is also often prescribed in primary APS patients but its efficacy is not proven. The initial management of VTE involves standard therapy with low molecular weight heparin followed by oral anticoagulation. Recurrent VTE events are managed using long term warfarin. The optimal intensity of long term anticoagulation must be determined balancing the risk of thrombosis against that of bleeding (1% per year, 0.25% severe haemorrhage).
In patients with stroke associated with aPL, management should involve long term warfarin. The optimal intensity remains disputed but in the absence of adequate data most adopt a conventional target international normalised ratio of 2.5, reserving higher intensity therapy for those with recurrent events. The addition of aspirin may also be of benefit but with increased risk of bleeding. In patients with stroke it is important to also correct conventional arteriopathic risk factors. The response of neurological syndromes to anticoagulation is variable, raising the question of the pathophysiological mechanism of dysfunction. New treatments such as rituximab29 have yet to be systematically evaluated in patients with neurological disorders.
THROMBOTIC THROMBOCYTOPENIC PURPURA
Thrombotic thrombocytopenic purpura (TTP) is a rare potentially fatal disorder characterised by red cell fragmentation (microangiopathy), haemolytic anaemia and thrombosis. This results in a consumptive thrombocytopenia and ischaemic organ injury. The development of such a prothrombotic milieu is a consequence of the presence of an abnormal number of unusually large Von Willebrand multimers (μl-VWF) in the circulation due to altered VWF homeostasis.30 VWF serves as the primary link between platelets and the subendothelium, and is normally released in response to prothrombotic stimuli which then undergoes proteolysis. The persistence of abnormally large VWF, as occurs in TTP, leads to platelet activation and clumping with subsequent microvascular thrombosis.
Central to the pathogenesis of TTP is the activity of a cleaving protease known as ADAMTS13. ADAMTS13 normally cleaves the μl-VWF into smaller peptides, but when the protease activity is reduced or absent, there is accumulation of excessive amounts of μl-VWF and the potential for TTP. ADAMTS13 activity can be reduced by several mechanisms with most cases of acquired TTP occurring in adults with autoantibodies against ADAMTS13. Congenital TTP presents in early life with ADAMTS13 deficiency in the absence of autoantibodies.
TTP occurs predominantly in adult women, usually occurring in association with infection, oestrogen containing preparations or pregnancy. Clinical features of TTP are diverse with the variability due to the presence of microthrombi in any number of organs and the degree of thrombocytopenia. Commonly, the symptoms at presentation are non-specific and include weakness, nausea, vomiting and abdominal pain. Fever is common as is renal impairment. Up to 50–75% of patients present with symptoms and signs consistent with overt cerebral ischaemia. This can range from strokes and transient ischaemic attacks to fluctuating neurological deficits. Headache, seizures, intermittent confusion and coma are frequently reported in acute TTP.
Laboratory evaluation of a suspected case of TTP is paramount. Thrombocytopenia and anaemia provide important clues. Careful evaluation of the blood film for red cell fragmentation may prove diagnostic (fig 1). Other laboratory parameters can also provide clues towards the diagnosis of TTP. Serum lactate dehydrogenase, bilirubin and reticulocytes are all elevated as a result of the concomitant haemolysis. Other forms of red cell fragmentation and haemolysis need to be excluded: disseminated intravascular coagulation, haemolytic uraemic syndrome, malignant hypertension, pre-eclampsia, HELLP (haemolysis, elevated liver enzymes and low platelets) and certain infections. Assays to assess ADAMTS13 activity and anti-ADAMTS13 antibodies have recently been developed and may be useful in the diagnosis and assessment of progress. Currently, these assays are a research tool and their role in clinical settings is being evaluated. Reduced ADAMTS13 activity alone is not pathognomic of TTP; low levels have also been found in disseminated intravascular coagulation, malignancy, uraemia, cirrhosis, acute inflammation and coronary syndromes.

Peripheral blood smear with red cell fragmentation.
The mainstay of therapy for acute TTP is intensive plasma exchange, which has reduced mortality rates from over 90% to 22%. This is attributed to the removal of ADAMTS13 autoantibody and replacement of ADAMTS13 activity by fresh frozen plasma or cryosupernatant. Immune suppression is usually coadministered with plasma exchange in the belief that it may result in a more durable response. In cases of refractory or relapsing disease, other immunosuppressive agents are often used and include ciclosporin, cyclophosphamide and vincristine. Recently, rituximab therapy has been associated with prompt remission and evaluation is ongoing.
Heparin induced thrombocytopenia
Heparin induced thrombocytopenia (HIT) is a potentially lethal complication of therapy with heparin, the most widely used parenteral medication in modern medicine. HIT occurs in approximately 3% of patients treated with unfractionated heparin, and less in patients receiving fractionated forms. The hallmark of HIT is the development of moderate thrombocytopenia and, despite the presence of heparin, a resultant prothrombotic state develops equating to a thrombotic risk 30 times that in control populations.31 This fundamental paradox of an anticoagulant becoming a procoagulant is caused by the development of antibodies against a self antigen which results in platelet activation. Heparin interacts with platelet factor 4, a platelet granular protein normally involved in haemostasis, immunoregulation and angiogenesis. This interaction results in a conformational change of PF4 and neoepitope exposure. Consequently, autoantibodies can develop which bind to the heparin–PF4 complexes and subsequently interact with platelets to cause platelet activation and thrombosis.
Thrombotic complications develop in 30–50% of cases. Venous thrombosis complicates HIT more often than arterial thrombosis, and pulmonary embolism is more common than all the arterial thrombotic events combined. The type and site of thrombosis varies according to the clinical setting; arterial thromboses are more frequent in those with cardiovascular disease and venous thromboses are more common in the postoperative patient. In more than 60% of cases, other concomitant prothrombotic risk factors exist, such as diabetes, neoplasm, cardiac insufficiency, systemic lupus erythematosus, APS, infection, trauma or indwelling lines (central venous catheters/arterial lines). Despite the thrombocytopenia, spontaneous bleeding is rare.
HIT must be rapidly recognised and if clinical suspicion is high, management involves immediate cessation of heparin and commencement of an alternative anticoagulant (direct thrombin inhibitors, heparinoids or danaparoid). Specialist haematologist input is required. Low molecular weight heparin should be avoided because of antibody cross reactivity, (between 50% and 80%), and warfarin monotherapy is initially contraindicated as it can promote venous gangrene.32 Unfortunately, the diagnosis is often elusive, complicated not only by the often featureless common clinical finding of thrombocytopenia, but by an array of laboratory tests that cannot independently diagnose HIT reliably. HIT is frequently described as a clinicopathological syndrome in which an accurate diagnosis requires a corroboration of selected clinical criteria with a positive laboratory test result (or pathological criterion). Laboratory testing in the diagnosis of HIT is based on either the immunological detection of antibodies directed against the PF4–heparin complex or the functional platelet activating potential of the emerging immunocomplexes.
SICKLE CELL DISEASE
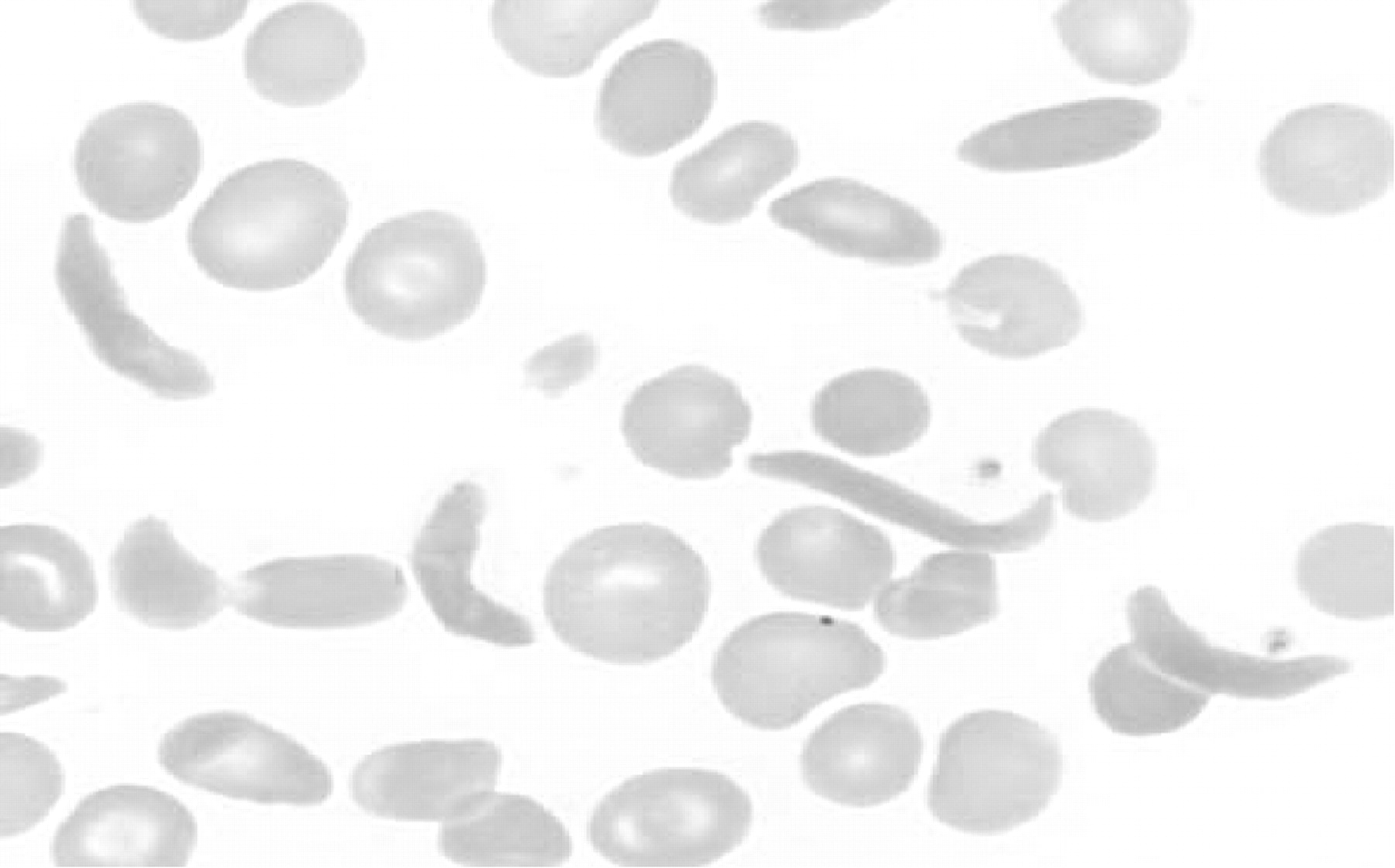
Sickle cell disease is a heterogeneous debilitating inherited disorder. The inherited abnormal gene is due to a point mutation on chromosome 11p15.5 which leads to an abnormal globin protein βs. Although the resultant sickle haemoglobin molecule can still retain the capacity to carry oxygen, it has the abnormal feature of being able to polymerise on deoxygenation and turn the normally pliable erythrocyte into a reversible but rigid elongated sickle shape (fig 2). This leads to haemolysis and the entrapment of sickled erythrocytes in the microcirculation. Over the past decade it has become clear that adhesive interactions between erythrocytes, other blood cells and endothelial cells add to the likelihood of vaso-occlusion.33 Furthermore, a proinflammatory state, hypercoagulability and endothelial dysfunction also play a role in the entrapment of sickle cells in the microcirculation.34

Peripheral blood smear showing sickled erythrocytes.
Carriers who inherit just one abnormal sickle gene (sickle cell trait, HbAS) are relatively asymptomatic compared with homozygotes who have sickle cell anaemia (HbSS) and are burdened with a range of complicated medical issues. In addition, more phenotypes exist in combination with other inherited haemoglobinopathies. Clinically, disease manifestations in sickle cell disease are attributed to either the haemolysis or vaso-occlusion. Red cell survival is only about 17 days in patients with HbSS compared with 120 days in people with normal haemoglobin. Despite tolerating their anaemia (tissue oxygenation is aided by a reduced affinity for oxygen by HbS), patients have a reduced oxygen capacity, develop a hyperdynamic circulation, an expanded plasma volume and dilated cardiomyopathy. Chronic haemolysis also leads to pigment gallstones and elevated levels of bilirubin.
The most frequent presentation of patients with sickle cell disease is due to the “painful crisis” caused by bone marrow ischaemia. Pain has a marked impact on quality of life in sickle cell patients. Involvement of the pulmonary circulation can result in the “acute chest syndrome” (occurs in up to 40% of patients), and chronic lung insults can result in both obstructive and restrictive lung disease, pulmonary hypertension and cor pulmonale. Hyposplenism renders the patient susceptible to infections with encapsulated organisms. Painful skin ulcerations around the ankle are also commonly seen and relate not only to vaso-occlusion of cutaneous microvasculature but also to other factors. Other commonly encountered problems include priapism, avascular necrosis of the femoral head, retinopathy, vitreous haemorrhage, hearing impairment and osteomyelitis.
Stroke is the leading cause of death in sickle cell and has the highest incidence in the first decade of life. Sickle cell disease is the most common cause of stroke in children.35 Ischaemic stroke due to middle cerebral artery stenosis/occlusion is common in the young whereas intracerebral or subarachnoid haemorrhage caused by Moya Moya-like friable collaterals predominantly affects adults. Lacunar syndromes also present to neurologists often when other conventional risk factors exist. Predisposition to stroke includes the HbSS phenotype, low Hb concentrations, leucocytosis, hypertension, previous acute chest syndrome, previous transient ischaemic attacks and a family history (increased risk in siblings suggesting a genetic factor). Silent brain disease with small ischaemic lesions may occur in up to one-third of patients and results in cognitive impairment.
Unfortunately, a cure for sickle cell disease remains elusive. Haemopoietic stem cell transplantation remains the only available potential curative therapy. Management of sickle cell disease needs a concerted team effort as damage to vital organs is greatly underestimated if one relies solely on clinical manifestations.36 Exchange blood transfusion remains the mainstay of therapy for the management of acute vaso-occlusive events in order to reduce the HbS%. Chronic red cell transfusion programmes are also employed for preventing major complications. Studies have shown that reducing the HbS% below 30% in patients with stroke significantly reduces the risk of recurrence.37 Primary prevention of stroke in children relies on recognition of those at high risk. Transcranial Doppler ultrasound has become a reliable tool for identifying those at risk by determining patients with high blood flow of major intracranial vessels. The STOP study demonstrated a marked reduction in risk of stoke in patients with high flow velocities who were subsequently managed with periodic transfusions to reduce the risk of stroke.38 Hydroxyurea is a relatively safe and effective adjuvant therapy.39 Hydroxyurea is a ribonucleotide reductase inhibitor which increases the fetal haemoglobin in sickle cell patients and is associated with fewer sickling events. However, up to 40% of patients may not respond well and because of side effects (myelosuppression, leg ulceration, potential malignant potential) is limited to patients with severe disease. New and developing agents for the treatment of sickle cell disease include anticellular adhesion therapies, omega-3 fatty acids, decitabine, valproate and erythrocyte ion channel blockers to prevent red cell dehydration.
PARAPROTEINAEMIC DISORDERS
The coexistence of neuropathy with a paraprotein is common and the relationship is often not causal. However, monoclonal gammopathy of undetermined significance (MGUS) is associated with a demyelinating neuropathy more often than by chance. The prototypic syndrome of distal acquired symmetrical demyelinating occurs in association with an IgM monoclone and anti-MAG antibodies.40 The same phenotype may be seen with IgG or IgA. Waldenstroms associated neuropathy is heterogenous and also associated with high titre IgM paraprotein. A causal relationship between a demyelinating neuropathy and a paraprotein is more likely when IgM paraprotein (MGUS or Waldenstroms) is associated with anti-MAG antibodies, or nerve biopsy shows complement or IgM deposition or widely spaced myelin. Axonal neuropathy is often present in patients with MGUS but the pathophysiology is often unclear. Other potential mechanisms of neuropathy with paraproteins include amyloid,41 cryoglobuinaemic,42 metabolic and other insults.
Multiple myeloma
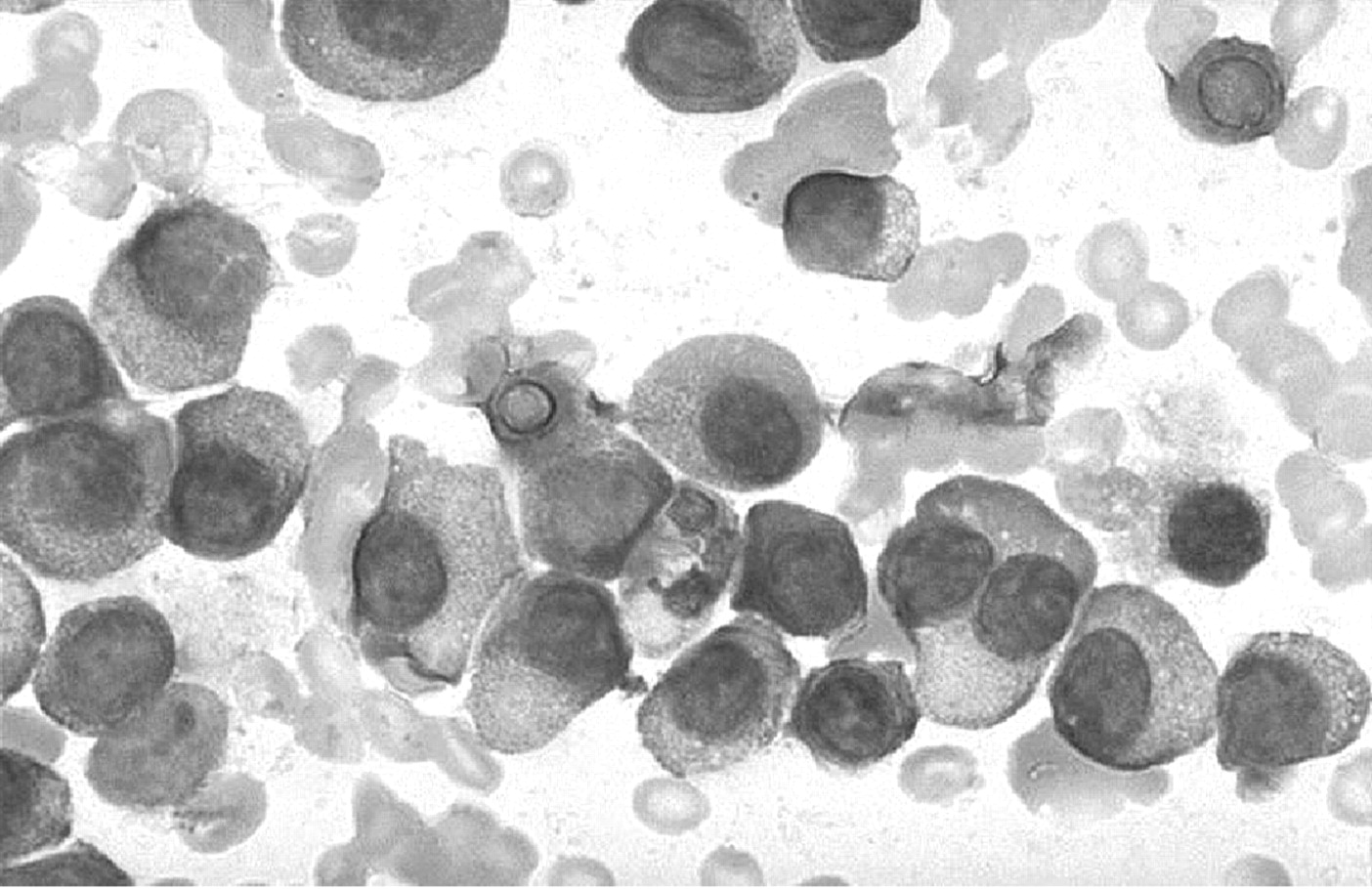
Multiple myeloma is a B cell neoplasm characterised by bony destruction and bone marrow replacement by a malignant clone of plasma cells that typically secrete monoclonal immunoglobulin (fig 3). Accounting for more than 10% of haematological malignancies, patients classically present with anaemia, renal failure and lytic bone lesions. Rapid progression occurs without treatment. Other complications include spinal cord compression, radiculopathy, bleeding, hyperviscosity syndrome, hyperuricaemia and hypercalcaemia. Despite advances in systemic and supportive therapies, multiple myeloma remains incurable because of chemotherapeutic resistance.
{kind=link}
{kind=link}
{kind=link}
A myeloma bone marrow aspirate with plasma cells.
Recent advances in this field have focused on the biology of the myeloma clone. Aside from the genetic alterations which cause overexpression of proto-oncogenes (common in many B cell malignancies), it is now understood that central to the pathogenesis of the tumour growth is the bone marrow microenvironment which supports the abnormal clone. The bone marrow microenvironment consists of cellular and extracellular components which interact with the plasma cells to promote growth and survival. Recent studies have shown that various growth factors, including interleukin (IL)-6, insulin-like growth factor, vascular endothelial growth factor and tumour necrosis factor family proteins play an important role in the pathogenesis and mediate cell proliferation, drug resistance and bone marrow migration.
High dose chemotherapy and bone marrow transplantation remain the treatments of choice for many patients although much interest has focused on developing new treatments that target not just the myeloma clone but the bone marrow microenvironment. Immunomodulatory drugs, including thalidomide, bortezomib and lenalidomide are now in regular therapeutic use, and combined with conventional therapies appear to enhance cytotoxicity and reduce drug resistance. Furthermore, novel agents that inhibit growth factor signalling cascades (by targeting ligands, receptors and/or downstream signalling cascade proteins) represent further promising therapeutic strategies.
Other plasma cell disorders
POEMS (osteosclerotic myeloma)
This is a rare plasma cell, multisystem disease, classically associated with sclerotic bone lesions and a chronic progressive neuropathy similar to chronic inflammatory demyelinating polyneuropathy. Nerve conduction studies often show both demyelinating and axonal features. The POEMS acronym refers to the well recognised features of the condition (Polyneuropathy, Organomegaly, Endocrinopathy, M Protein, and Skin changes) although it is rare to have all five features. Classic features of myeloma are usually not present, and the size of the paraprotein is only small.
The cause of the paraneoplastic-like processes associated with POEMS is not well understood. These patients often have higher levels of growth factors (IL-6, tumour necrosis factor, IL-1, vascular endothelial growth factor) than in myeloma but do not demonstrate immunoglobulin deposition in the tissues.
Unlike, myeloma, POEMS is a chronic disorder with a median survival of 14 years. Nevertheless, quality of life is poor and patients can die of multiorgan failure or other complications. Sclerotic lesions often respond to radiotherapy but unlike other chronic demyelinating polyneuropathies, plasmapheresis and/or intravenous immunoglobulin therapy does not improve the neurological symptoms. Treatment is currently limited to immunosuppression or chemotherapy. High dose chemotherapy with stem cell rescue is an option in the younger patient. Recent experience with four POEM patients undertaking an autologous stem cell transplant with high dose melphalan revealed a significant improvement in organ disease and a corresponding reduction in vascular endothelial growth factor levels.43
Monoclonal gammopathy of undetermined significance
MGUS is characterised by the presence of an elevated paraprotein level without any evidence of another plasma cell disorder or lymphoproliferative disease. It is a common finding (1% in ages >50 years, 3% in ages >70 years), with a relatively good prognosis although approximately 1–3% of MGUS cases per year progress into an aggressive plasma cell dyscrasia or lymphoproliferative disease. Determinants for the transformation of MGUS to myeloma are unknown although studies suggest that the paraprotein level at diagnosis is the most important predictor of progression.44
Management of patients with MGUS centres on periodic clinical evaluation and paraprotein determination in order to screen for any progression to a more aggressive plasma cell dyscrasia, or the development of other related conditions (table 3). MGUS is associated with other disorders, including connective tissue disorders, other haematological diseases and neuropathy.
Investigations for suspected monoclonal gammopathy of undetermined significance
Waldenströms macroglobulinaemia
Waldenströms macroglobulinaemia (WM) is a neoplasm of small B lymphocytes, plasmacytoid lymphocytes and plasma cells, usually involving bone marrow, lymph nodes and spleen. It is characterised by a monoclonal IgM paraprotein, with hyperviscosity or cryoglobulinaemia a common complication. WM is relatively rare, accounting for approximately 2% of all haematological malignancies. Unlike many lymphoproliferative diseases, and more in keeping with plasma cell diseases, there is a lack of a defining genetic hallmark (deletion of 6q and translocation of chromosomes 9 and 14 [t(9;14)] are the most common findings). The postulated cell of origin is the peripheral blood lymphocyte simulated to differentiate into a plasma cell.
Most patients with a diagnosis of WM have symptoms attributable to tumour infiltration and/or monoclonal protein. Infiltration of a variety of organs can occur. CNS involvement is rare but neuropathy is common and may be the presenting feature. Deposition of IgM into tissues can cause glomerular damage, skin nodules and malabsorption. Amyloid deposition has also been reported in WM. Median survival time of patients with WM ranges between 5 and 10 years, with age, B2-microglobulin and degree of anaemia being important prognostic factors for survival.45
The aim of treatment for patients with WM is to improve the quality and duration of life with minimal side effects. It is not yet clear if achievement of a complete remission confers clinical benefit because of increased toxicity of treatment. Plasmapheresis is indicated for the acute management of patients with severe problems due to a circulating paraprotein. Despite a lack of comparative data, alkylating agent-based treatments, combination therapy or purine analogues are all suitable choices for initial therapy of patients requiring treatment. Autologous stem cell transplantation may have a role in selected patients, with reports in the literature to suggest that this is a relatively safe, feasible option associated with significant cytoreduction. The use of allogeneic transplantation in WM has also been reported, with some success.46 In younger patients, in whom high dose treatment is contemplated, there is a role for the use of monoclonal antibody therapy. Monoclonal antibodies targeting CD20, CD22 and CD52 have been successfully used in B cell malignancies. WM cells nearly always have CD20 antigen, and the use of an anti-CD20 antibody (rituximab) and other monoclonal antibodies has produced good response rates.47 Newer therapies include interferon α, thalidomide, bortezomib, oblimersen sodium and sildenafil. Further developments in cell and molecular biology are likely to help in the development of targeted therapies for WM.
Table 4 provides a summary of all the recent advances in the haematological conditions discussed in this article.
Summary of the recent advances in haematological conditions
REFERENCES
Footnotes
-
Competing interests: None.