Article Text

Abstract
Background Some patients meeting behavioural variant frontotemporal dementia (bvFTD) diagnostic criteria progress slowly and plateau at mild symptom severity. Such patients have mild neuropsychological and functional impairments, lack characteristic bvFTD brain atrophy and have thus been referred to as bvFTD ‘phenocopies’ or slowly progressive (bvFTD-SP). The few patients with bvFTD-SP that have been studied at autopsy have demonstrated no evidence of FTD pathology, suggesting that bvFTD-SP is neuropathologically distinct from other forms of FTD. Here, two patients with bvFTD-SP with chromosome 9 open reading frame 72 (C9ORF72) hexanucleotide expansions are described.
Methods 384 patients with an FTD clinical spectrum and Alzheimer's disease diagnoses were screened for C9ORF72 expansion. Two bvFTD-SP mutation carriers were identified. Neuropsychological and functional data, as well as brain atrophy patterns, assessed using voxel based morphometry (VBM), were compared with 44 patients with sporadic bvFTD and 85 healthy controls.
Results Both patients were aged 48 years at baseline and met possible bvFTD criteria. In the first patient, VBM revealed thalamic and posterior insula atrophy. Over 7 years, his neuropsychological performance and brain atrophy remained stable. In the second patient, VBM revealed cortical atrophy with subtle frontal and insular volume loss. Over 2 years, her neuropsychological and functional scores as well as brain atrophy remained stable.
Conclusions C9ORF72 mutations can present with a bvFTD-SP phenotype. Some bvFTD-SP patients may have neurodegenerative pathology, and C9ORF72 mutations should be considered in patients with bvFTD-SP and a family history of dementia or motor neuron disease.
Statistics from Altmetric.com
Introduction
Behavioural variant frontotemporal dementia (bvFTD) is a neurodegenerative clinical syndrome characterised by insidious behavioural and personality changes. One lingering challenge to bvFTD diagnostic criteria1 is that some patients meeting criteria present with a slowly progressive course (bvFTD-SP) and plateau at mild symptom severity.2 These patients have been referred to as bvFTD ‘phenocopies’ because, although their behavioural features resemble bvFTD, they do not display typical patterns of brain atrophy or hypometabolism at baseline,3 4 nor do they show progressive volume loss typical of bvFTD, leading some authors to suggest that the syndrome is not caused by a neurodegenerative disease.1 5 6 The few individuals who have been examined with brain autopsy did not show FTLD pathology,1 and because these patients often have normal life spans,1 large pathological series are not available, leaving the neuropathological correlates of bvFTD-SP unclear.
Despite estimates from one group that ‘phenocopies’ may account for one-third of patients with bvFTD,2 there has yet to be a study linking bvFTD-SP to a known FTD associated mutation. Recently, a hexanucleotide expansion in chromosome 9 open reading frame 72 (C9ORF72) was shown to be the most common genetic abnormality in both familial and sporadic bvFTD and amyotrophic lateral sclerosis (ALS), with bvFTD being the major phenotype in carriers with a dementia syndrome.7 8 All patients examined displayed frontotemporal lobar degeneration with TDP-43 positive inclusions (FTLD-TDP) pathology.9 10 Here we report two patients who were diagnosed with bvFTD-SP at the University of California, San Francisco (UCSF) and had C9ORF72 expansions. The presence of the C9ORF72 (C9FTD/ALS) mutation in these patients suggests that some patients with bvFTD-SP may have slowly progressive FTD with pathological inclusions containing TDP-43, the protein associated with bvFTD and ALS in C9ORF72 mutation carriers.7 8
Methods
Participant selection
Three hundred and eighty-four patients with FTD clinical spectrum diagnoses and possible or probable Alzheimer's disease (AD) diagnoses (National Institute of Neurological Disorders and Stroke–Alzheimer's Disease and Related Disorder Association research criteria) were tested for C9ORF72 expansion (figure 1). Eighty-seven patients had a diagnosis of bvFTD (n=61) or FTD-ALS (n=26). Twenty-three (bvFTD=13; FTD-ALS=10) of the 87 patients carried the mutation (C9+). No patient with a non-bvFTD clinical syndrome was C9+. Four patients (2 C9+, 2 C9−) were identified as bvFTD-SP. Patients were identified as bvFTD-SP if they met the following criteria: (1) symptomatic for ≥5 years, (2) met FTD Consortium (FTDC) international criteria for bvFTD (possible or probable),11 (3) no decline on the Clinical Dementia Rating Sum of Boxes (CDR-SB) score over at least 2 years after initial evaluation and (4) little or no atrophy on visual assessment of structural MRI at the most recent evaluation. The follow-up period in criterion 3 was used in a previous study by another group to identify bvFTD-SP.2 Demographic and non-neurological clinical data have been modified to maintain anonymity. All study participants (or their surrogates) provided written informed consent, and all study procedures were approved by local institutional review board.

Diagnoses of patients screened for the C9ORF72 mutation. AD, Alzheimer's disease; ALS, amyotrophic lateral sclerosis; bvFTD, behavioural variant frontotemporal dementia; FTD, frontotemporal dementia; SP, slowly progressive.
Genotyping
The GGGGCC hexanucleotide repeat expansion in C9ORF72 was detected using a two step PCR based protocol, as previously described.7 Briefly, the hexanucleotide repeat was amplified in all samples using one fluorescently labelled PCR primer. Next, fragment length analysis was performed on an automated ABI3730 DNA analyzer. All patients that appeared homozygous in this assay were next analysed using a repeat primed PCR method where characteristic stutter amplification pattern on electropherogram was considered evidence of pathogenic C9ORF72 expansion.
Imaging
Each patient underwent structural MR scanning on a 1.5 T (2005–2006), 3 T (2009–2011) or 4 T (2007–2008), and images were preprocessed for voxel based morphometry (VBM) using DARTEL running in SPM5 with Matlab V.7.7 (scanner protocols and preprocessing have been described previously).12 Single subject VBM of combined grey–white matter images for each patient's initial scan were compared with 50 healthy controls selected to match each patient in age (within a decade) and scanner type. Because single subject VBM typically lacks statistical power to detect regions of clinically significant atrophy, unthresholded maps were generated to depict the full extent of each patient's atrophy relative to controls. For comparative purposes, maps were also generated using 20 patients with C9− bvFTD and 164 healthy controls. Positron emission tomography (PET) with the β-amyloid ligand [11C]PIB and [18F]-labelled fluorodeoxyglucose (FDG) was performed, using previously described methods,13 on one of the C9+ patients with bvFTD-SP.
Neuropsychological and social–emotional testing
Each patient was tested using a previously described neuropsychological examination (table 1).14 For comparative purposes, neuropsychological data from 44 C9− patients with typical bvFTD (aged 61.8±6.6 years (range 46–83); education 16.1±2.4 years; gender 36.3% female; CDR 1.1±0.5 (range 0.5–2); CDR-SB 6.8±2.8 (range 1.5–14); Functional Activities Questionnaire (FAQ) score 17.8±7.6) and 85 cognitively normal middle aged controls are shown (age range 50–64 years; education 17.8±2.6 years; 50 women/35 men). Clinically significant impairment (performance less than the fifth percentile relative to control subjects) was identified by converting patient raw scores to Z scores. Social–emotional functioning was quantified using previously described methods.15
Performance on neuropsychological measures
Case reports
Patient No 1
Patient No 1 was a 48-year-old right-handed man referred for evaluation after a 5 year history of personality change, memory and visuospatial dysfunction, and an inability to work. His first symptoms were misplacing objects, getting lost in familiar neighbourhoods and forgetting song lyrics. Soon thereafter, he experienced spells during which he would become unresponsive and uncooperative for hours, several times per week. At night, he would walk through his home aimlessly, resisting re-direction from his wife. The patient became progressively more rigid regarding dietary preferences, eating fixed meals and requiring his family to adopt similar dietary restrictions. After being fired from his job for violating protocols, he developed severe anxiety and depression, and attempted suicide.
The patient then sought medical attention. He was first diagnosed with post-traumatic stress disorder (PTSD) and depression, and later with bipolar disorder type II. Although psychiatric medications were effective for many of his psychiatric symptoms, he developed increasing memory difficulties; he began to lose his train of thought mid-sentence and developed word finding difficulty. Previously an involved father, the patient was indifferent when his child was hospitalised, and became increasingly unsympathetic towards friends and family. He became compulsively fixated on computer games and began to have delusions about his family, accusing them of lying to him. These changes prompted an evaluation at our centre.
Patient No 1 reported a lifetime of dysthymia and superimposed major depression. He had no history of head injury or loss of consciousness and had never taken acetylcholinesterase inhibitors or memantine. He obtained a Master's degree and worked successfully as a mortician for 20 years. There was no history of alcohol, tobacco or recreational drug abuse. His maternal family history was remarkable for a grandmother diagnosed with AD at an unknown age and an aunt with obsessive–compulsive disorder. His paternal side was significant for late life AD in his grandmother, an aunt diagnosed with late life schizophrenia in her mid-40s and later AD in her 60s, an uncle and cousin diagnosed with bipolar affective disorder and another cousin with unspecified psychiatric illness (see supplemental figure 1, available online only). The patient's father had a progressive cognitive, behavioural and motor disorder that progressed to death at age 73 years. Autopsy was performed, and follow-up immunohistochemistry was performed on archival tissue blocks from frontal cortex and medial temporal lobe. These studies revealed an unclassifiable subtype of FTLD-TDP with TDP-43 immunoreactive dots, threads and neuronal cytoplasmic inclusions that were sparse in the frontal and entorhinal cortex and moderate in the hippocampal CA1/subiculum, accompanied by hippocampal sclerosis (further described in the supplementary data, available online only).
On examination, the patient was alert, oriented and well groomed but had a flat affect. He was overly familiar, perseverative, angry and critical of others throughout the interview. He frequently interrupted the examiner and raised his voice inappropriately. He showed poor insight into his symptoms, making excuses for his deficits. Cranial nerve examination revealed mild flattening of the right nasolabial fold with normal facial strength. Motor examination was normal for bulk and power but there was mild axial rigidity. Stride length was normal but arm swing was diminished on the right greater than the left. He had mild retropulsive instability.
On neuropsychological evaluation14 (table 1) the patient scored 30/30 on the Mini-Mental State examination16 and performed normally on verbal episodic memory testing, speech and language, visuoconstruction, visuoperceptual function and calculations. He performed below expectations on visual memory free recall but his recognition was normal. On measures of executive function, he scored in the impaired range relative to matched controls on phonemic fluency, category fluency and cognitive control (Stroop) tasks, and his working memory (digits backward) was below expectations although he was able to complete a set shifting and sequencing task rapidly and accurately. He endorsed 20 items on the Geriatric Depression Scale (GDS),17 consistent with clinically significant depression. His Neuropsychiatric Inventory18 total score was 61, and was notable for elevated agitation, anxiety, depression, disinhibition, irritability and aberrant eating behaviour. The patient scored 11 on the FAQ,19 suggesting mild impairment in activities of daily living, and his global CDR20 score was 1, with Sum of Boxes equal to 5 (table 2), suggestive of very mild functional impairment.
Disease severity and functional impairment
Social–emotional testing revealed pathologically low levels of empathy, characterised by an inability to think about others' emotions (Interpersonal Reactivity Index–Perspective Taking (IRI-PT) 12/35) and irritation from others' emotional expressions (IRI-Personal Distress (PD) 23/35) although his level of empathic concern was not in the impaired range (IRI-Empathic Concern (EC) 24/35).21 He was pathologically insensitive to subtle social cues (Revised Self-Monitoring Scale (RSMS) 37/84)22 but was sensitive to perceived criticism and afraid of making errors (Behavioural Inhibition Scale (BIS) 27/28).23 He was impaired at naming emotions (The Awareness of Social Inference Test-Emotion Evaluation Test (TASIT-EET) 9/14)24 although he could detect sarcasm (TASIT-Social Inference Minimal (SIM): Simple Sarcasm (SSR) 19/20, Paradoxical Sarcasm (PSR) 17/20). His emotional theory of mind was impaired (Castelli Triangles Intentionality 11/20).25 Personality ratings through the Interpersonal Adjectives Scale indicated impaired warmth (T=32), and pathologically high levels of arrogance (T=81) and dominance (T=78) were a part of the patient's normal personality before becoming ill that had further intensified since symptom onset.26
A structural brain MRI revealed no abnormalities or gross atrophy, as rated visually by UCSF clinicians (figure 2A). At age 51 years the patient also underwent FDG-PET that was within normal limits based on qualitative and quantitative assessments. A PET scan with PIB did not reveal evidence of cortical amyloid. CSF analysis revealed normal τ and A-β levels, not suggestive of AD. An EEG was unremarkable.
{kind=link}
{kind=link}
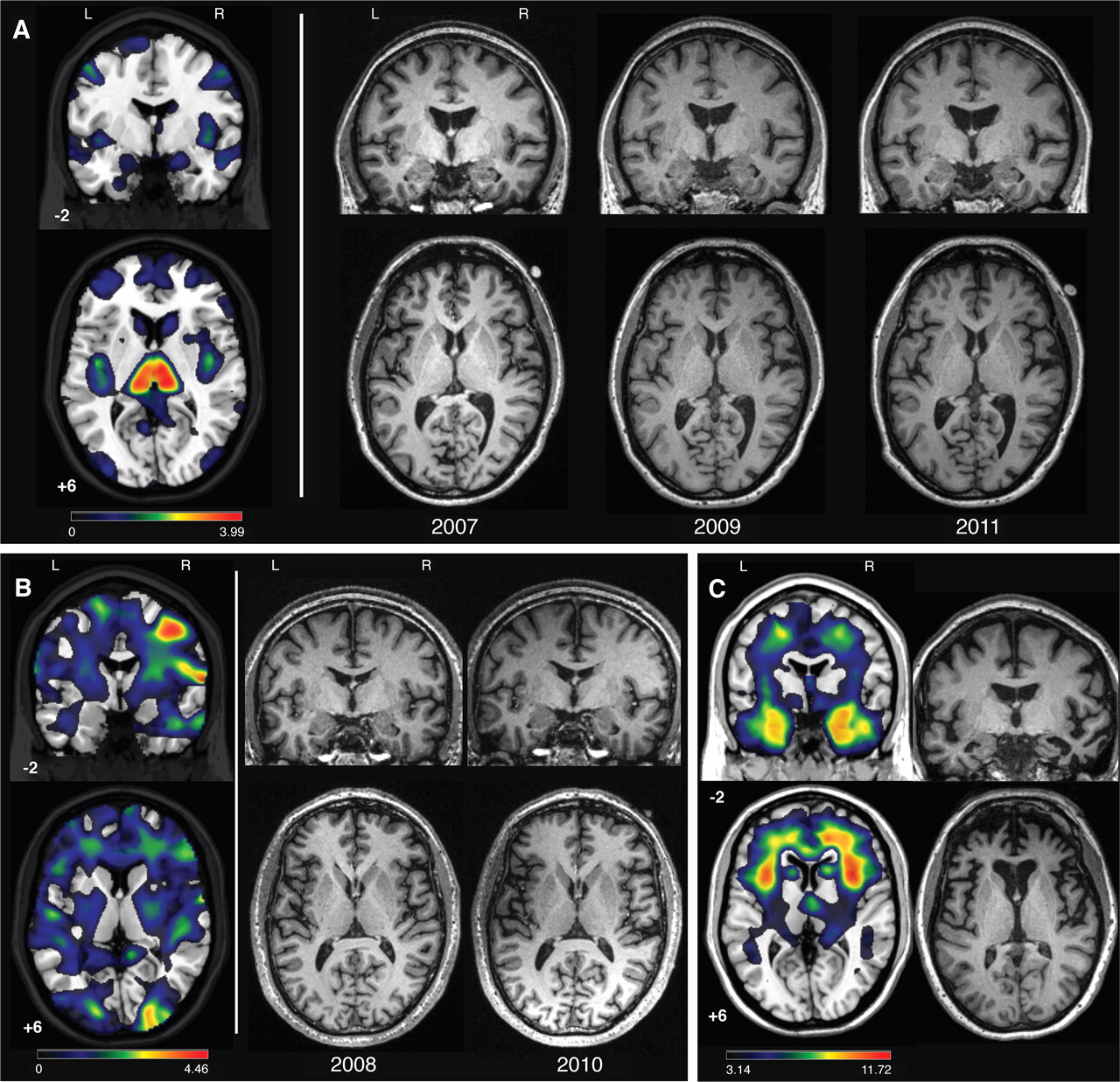
Patient Nos 1 and 2 single subject voxel based morphometry (VBM) and serial MRIs, and typical behavioural variant frontotemporal dementia (bvFTD) atrophy pattern. (A) Patient No 1: single subject VBM for patient No 1 using the second (2009) scan is shown in the left panel with unthresholded T map (0<T<3.99) results overlaid on the template brain. On the right, slices from three serial structural T1 MRIs are provided for patient No 1 from 2007, 2009 and 2011. (B) Patient No 2: single subject VBM results with an unthresholded T map (0<T<4.46) are shown overlaid on the template brain in the left panel, with corresponding slices from patient No 2′s structural MRIs from 2008 to 2010 shown in the right panel. VBM was performed on the 2010 image. (C) Typical bvFTD atrophy pattern: VBM results comparing 20 bvFTD with 164 cognitively normal controls overlaid on the template brain are shown on the left (p<0.001). On the right is a single demographically matched bvFTD patient's structural MRI. MNI coordinates are provided for template images (template brain in MRIcron: ch2.nii), and for all coronal and axial slices the left side of the image represents the left side of the patient. This figure is produced in colour in the online journal—please visit the website to view the colour figure.
The patient was evaluated annually for the next 5 years. During this period, he developed new compulsions (eg, repetitively watching grotesque horror films) and had increasing loss of empathy towards family and friends. On social–emotional testing, he became less able to recognise socially inappropriate behaviours, could no longer detect sarcasm or deception in others, and his moral reasoning became highly utilitarian and emotionally detached. In other domains the patient remained stable. His neurological examination, neuropsychological scores (table 1) and MRI (figure 2A) remained unchanged. Some memory scores improved (table 1) but this was likely the result of a practice effect. Notably, the patient's CDR and FAQ scores improved slightly with longitudinal follow-up (table 2). Moreover, his clinicians noted in subsequent visits that the patient displayed greater insight into his disease, learning to compensate for his cognitive and behavioural changes. For example, he adapted to his memory deficits by becoming more organised and regimented, and developed a list of social rules to guide his conduct when interacting with others.
Based on the patient's initially progressive symptoms, he met FTDC criteria11 at the initial evaluation. Due to his stable neurological, neuropsychological and imaging tests at later evaluations, he met the present criteria for bvFTD-SP. Single case VBM compared with normal controls revealed bilateral thalamic and mild posterior insular atrophy (figure 2A).
Patient No 2
Patient No 2 was a 48-year-old right-handed woman who reported a 5 year history of behavioural and personality changes. Her symptoms began when she became irritable, self-centred and emotionally detached. For example, she refused to visit her son in the hospital after he had major surgery, and she was resentful and angry during her son's recovery period. She displayed inappropriate behaviours such as sneaking up on a coworker from behind, pretending to have a gun. Previously flexible in her daily routine, the patient began to ritualistically follow a rigid daily schedule with set waking, sleeping and eating times, and she would get angry if this routine was broken. At age 46 years she sought medical evaluation due to her increasing confusion and distractibility. She was diagnosed with bvFTD by an outside neurologist despite a reportedly normal MRI. Over the next 2 years she became depressed, her speech became more tangential and she gradually lost her ability to use simple household appliances.
Patient No 2's medical history was significant only for hypertension. She had a history of dyslexia. She completed 10th grade and worked as an exterminator until her late 20s before managing a construction company until becoming ill. The patient endorsed alcohol and drug abuse in her 20s, including cocaine and marijuana. At the time of her evaluation, the patient had stopped drinking alcohol but continued to smoke marijuana. Both of her parents had a history of alcohol abuse, and late life behavioural disorders were reported in her father and an older brother.
On examination, the patient was pleasant and cooperative but occasionally became agitated when discussing her symptoms. Her speech was fluent with rare phonemic paraphasic errors. Naming and comprehension were normal. Neurological examination was otherwise unremarkable. On neuropsychological testing (table 1), the patient scored 23/30 on the Mini-Mental Status Examination (losing points for orientation, recall, repetition, visuospatial function and following a command). Verbal episodic learning and delayed free recall were poor, but recognition was normal, and visuospatial episodic learning and recall were normal. Language and visuospatial functions were normal. She was impaired in calculations due to careless errors rather than frank acalculia. She was impaired on lexical fluency, was slow on a set shifting task, and her abstraction and working memory were below expectations. Her design fluency and category fluency were normal. She endorsed 9/30 items on the GDS. Neuropsychiatric Inventory total score was 35 and was notable for delusions, agitation, anxiety, euphoria, apathy, disinhibition and eating changes. The patient scored 20 on the FAQ and her global CDR score was 1, with Sum of Boxes equalling 7.5 (table 2).
Social–emotional testing showed impaired ability to think about others' emotions (IRI-PT 14/35), extreme irritation by others' emotions (IRI-PD 33/35) but low–normal empathic concern (IRI-EC 23/35). She was pathologically insensitive to subtle social cues (RSMS 30/84) but showed high average sensitivity to criticism (BIS 20/28). She was impaired at naming emotions (TASIT-EET 9/14) and recognising sarcasm (TASIT-SIM: SSR 13/20; PSR 14/20). Compared with informant ratings of her premorbid personality (NEO-PI-3),27 she had significantly increased neuroticism, including anxiety (+8), depression (+10), angry hostility (+7) and self-consciousness (+10), along with decreased extraversion and more mild decreases in agreeableness.
The patient's brain MRI at initial evaluation revealed no gross atrophy or abnormalities by visual inspection, and 2 years later her scan remained unchanged (figure 2B). Single case VBM compared with normal controls revealed a non-specific pattern of parieto-occipital atrophy with more subtle frontal and insular changes (figure 2B).
During the two intervening years her husband noted slightly increased apathy but otherwise her symptoms remained stable and her neurological and neuropsychological assessments remained unchanged (table 1). In fact, her CDR-SB score decreased (table 2). Compared with her initial visit, the patient demonstrated greater insight into her illness, showing concern over her symptoms and prognosis, and personality testing showed decreased neuroticism. She continued to be impaired on tests of emotion naming and empathy but her performance did not worsen. The patient's progressive behavioural and personality changes fulfilled FTDC criteria at the initial evaluation. Due to stable symptoms without characteristic bvFTD brain atrophy, she was diagnosed with a slowly progressive variant.
Discussion
We have reported two patients diagnosed with a slowly progressive bvFTD (bvFTD-SP), or ‘FTD phenocopy’, who were subsequently found to carry the C9ORF72 expansion. These individuals initially fulfilled international criteria for possible bvFTD and were later diagnosed with bvFTD-SP due to their stable clinical course over 7 (patient No 1) and 2 (patient No 2) years of follow-up and imaging uncharacteristic of bvFTD. These findings demonstrate that a subset of patients with bvFTD-SP may have neurodegeneration due to underlying FTLD-TDP pathology. The presence of slowly progressive forms of C9+ bvFTD may reflect a difference in the type of underlying C9ORF72 mutation (eg, number of hexanucleotide repeats), modifying effects of genetic/epigenetic factors or incomplete penetrance of the allele. As the overall number of bvFTD-SP patients was small in our C9ORF72 expansion screened FTD population, it is not possible to reliably estimate the prevalence of C9+ bvFTD-SP; however, both patients had a family history of dementia. Our findings therefore suggest that C9ORF72 mutations should be entertained in patients with bvFTD-SP, particularly if there is a known family history of dementia.
To date, only a handful of bvFTD-SP patients have come to autopsy, and none was found to harbour FTLD pathology.1 Structural imaging changes are not mandatory for a definite bvFTD diagnosis if a known genetic cause of FTD is present,11 and the lack of such atrophy over longitudinal assessment makes the diagnosis of neurodegenerative disease uncertain.6 As assessed visually by our clinicians, neither patient had a typical bvFTD atrophy pattern on initial visit or on longitudinal structural MRIs (figure 2). Accordingly, these patients would have received a possible bvFTD diagnosis using FTDC international research criteria. Patient No 1's normal FDG-PET is consistent with reports of normal FDG-PET in other bvFTD-SP patients.3 Retrospectively, VBM revealed thalamic and posterior insular atrophy in patient No 1 and a non-specific pattern of parieto-occipital, frontal and insular atrophy in patient No 2; neither of these patterns was deemed clinically significant by the treating physicians at the time of evaluation. While the full spectrum of imaging abnormalities associated with C9FTD/ALS has not yet been described, the pattern of atrophy observed in patient No 2 is similar to what we have described in the VSM-20 C9FTD/ALS family in whom the C9ORF72 mutation was first identified.7 9 These findings raise the possibility that C9FTD/ALS associated brain atrophy patterns could be used to identify slowly progressive C9ORF72 mutation carriers with a bvFTD-SP phenotype.
Prominent frontal and insular atrophy is a consistent neuroanatomical feature of bvFTD. VBM analysis of patient No 1 showed no frontal atrophy while insular atrophy was more posterior than typically found in patients with bvFTD (figure 2A–C). While VBM in patient No 2 revealed frontal and insular atrophy, compared with other patients with bvFTD, these changes were unrecognised on visual inspection of structural imaging and were only detected using single subject VBM (figure 2B). The lack of significant atrophy in characteristic bvFTD regions may explain why both patients maintained a level of insight into their illness. Anosognosia, or the lack of awareness into one's deficits, has been linked to atrophy in the frontal lobes,28 and the lack of significant frontal atrophy in both patients may be why both patients better adapted to their deficits. Patient No 1 eventually developed strategies to cope with his symptoms, and during follow-up evaluation, patient No 2 was actively engaged with questions about her disease and interest in clinical trials. Interestingly, patient No 1 generated a list of rules to guide his conduct during social situations. While this demonstrates a depth of insight into his illness, it is also reflective of the obsessive features frequently observed in bvFTD.
Similarly, although both patients showed many deficits on socioemotional testing, such as impaired emotion naming and difficulty imagining others' thoughts and emotions, they both retained a rudimentary concern for others and themselves that is atypical of bvFTD. Greater insight and some preservation of basic emotional responding could also be the reason that both individuals developed significant depression and anxiety, consistent with reports that individuals with bvFTD-SP may have higher rates of depression compared with typical bvFTD.2 In fact, both patients reported more items on the GDS across longitudinal evaluations, and at the time of the final evaluation had higher GDS scores than other bvFTD patients (table 1).
Despite the lack of frontal atrophy, both patients displayed executive dysfunction on neuropsychological testing. While impaired on some neuropsychological tests, both patients outperformed individuals with typical bvFTD on many of the executive tasks (table 1), consistent with reports that patients with bvFTD-SP can be differentiated from progressive bvFTD through such tests.1 Moreover, given that both patients were at least 7 years into their disease course at the final evaluation and that time from first symptom to death averages 6 years,29 these patients' impairments were remarkably mild for typical bvFTD. Importantly, mild executive dysfunction is not unusual in bvFTD, and 25% of patients with typical bvFTD initially perform within normal limits on standard neuropsychological testing.30 Thus normal performance on cognitive testing may be supportive of a diagnosis of bvFTD-SP, but it does not rule out typical bvFTD.
Taken together, our findings suggest that clinicians should strongly consider FTLD pathology in patients who meet criteria for bvFTD-SP, particularly when there is significant psychiatric comorbidity, as was the case in patient No 1. Up to 51% of patients with bvFTD receive erroneous diagnoses at initial evaluation due to symptoms that resemble psychiatric diseases.31 Patient No 1 was initially diagnosed with post-traumatic stress disorder and bipolar affective disorder due to his early symptoms. The presence of prominent psychiatric illness in the families of both patients may further indicate underlying neurodegenerative disease,31 yet the absence of such history in bvFTD-SP would not rule out C9+ bvFTD, since sporadic patients with C9ORF72 bvFTD are not uncommon.7 Notably, the limited materials available from the father's autopsy suggest an unclassifiable form of FTLD-TDP as well as ubiquitin positive, TDP-43 negative neuronal cytoplasmic inclusions in the medial temporal lobe; these features strongly suggest a C9ORF72 linked illness.32
One of the most enigmatic and diagnostically challenging features of patients with bvFTD-SP is their altered personality and behaviour in light of unremarkable imaging assessments.6 A possible explanation for the behavioural symptoms we observed in our subjects is that these features derive from neuronal and synaptic dysfunction that is not well captured on standard neuroimaging assessments. It is possible that newer imaging techniques such as intrinsic connectivity functional MRI may be more sensitive to such C9ORF72 related brain dysfunction.33 34 It will also be of interest to continue to monitor these patients for the emergence of more typical bvFTD imaging abnormalities.
There are limitations to this report. In our cohort of 61 patients with bvFTD without comorbid motor neuron disease who were screened for the C9ORF72 variant, only four patients had been diagnosed as bvFTD-SP, which is lower than the frequency of bvFTD-SP reported by others (26 of 71 bvFTD in one series).2 This difference may be due to the limited longitudinal data available within our bvFTD cohort (31 out of 61 patients were followed for ≥2 years). However, we can accurately report the frequency of bvFTD-SP due to C9ORF72 expansion to be 15% in our cohort of 13 C9+ carriers. None of the other 11 carriers with bvFTD had bvFTD-SP; seven had progressive changes on longitudinal evaluation and all had significant atrophy at the first evaluation. Nevertheless, larger studies are needed to verify this estimate as our cohort of C9ORF72 carriers was small.
Here we have identified a novel link between bvFTD-SP and a known FTD causing mutation. Our findings suggest that an underlying neurodegenerative aetiology should not be ruled out in patients who meet criteria for possible bvFTD but have atypical features consistent with a bvFTD ‘phenocopy’. We also speculate that screening larger series of bvFTD-SP will likely identify other patients with C9ORF72 mutations.
Acknowledgments
The authors thank Dr William Jagust for PET imaging data. We thank our patients and their families for participating in neurodegenerative disease research.
References
Footnotes
See ALS and FTD Special Edition, p 355
Funding This work was supported by National Institutes of Health (grants R01AG031278, R01 AG038791, R01NS065782, R01 AG026251, R01 AG026938, R01 AG029577, K23 AG031861, K23 AG021606, P01 AG19724, M01-RR0079, P50 AG023501 and P50 AG1657303); the Hellman Foundation, the Tau Research Consortium, the ALS Association (ALSA), the State of California, Alzheimer's Disease Research Center of California (ARCC) (grant 03-7527) and the Larry Hillblom Foundation grants 2002/2J and 2007/2I.
Competing interests RR has a patent pending for the C9ORF72 non-coding expansion.
Ethics approval Ethics approval was provided by UCSF institutional review board.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- ALS and FTD Special Edition