Article Text

Abstract
Objectives To determine the safety and efficacy of lithium for the treatment of amyotrophic lateral sclerosis (ALS) in a randomised, placebo controlled, double blind, sequential trial.
Methods Between November 2008 and June 2011, 133 patients were randomised to receive lithium carbonate (target blood level 0.4–0.8 mEq/l) or placebo as add-on treatment with riluzole. The primary endpoint was survival, defined as death, tracheostomal ventilation or non-invasive ventilation for more than 16 h/day. Secondary outcome measures consisted of the revised ALS Functional Rating Scale and forced vital capacity. Analysis was by intention to treat and according to a sequential trial design.
Results 61 patients reached a primary endpoint, 33 of 66 in the lithium group and 28 of 67 patients in the placebo group. Lithium did not significantly affect survival (cumulative survival probability of 0.73 in the lithium group (95% CI 0.63 to 0.86) vs 0.75 in the placebo group (95% CI 0.65 to 0.87) at 12 months and 0.62 in the lithium group (95% CI 0.50 to 0.76) vs 0.67 in the placebo group (95% CI 0.56 to 0.81) at 16 months). Secondary outcome measures did not differ between treatment groups. No major safety concerns were encountered.
Conclusions This trial, designed to detect a modest effect of lithium, did not demonstrate any beneficial effect on either survival or functional decline in patients with ALS.
Trial registration number NTR1448. Name of trial registry: Lithium trial in ALS.
Statistics from Altmetric.com
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disorder characterised by progressive weakness. ALS is caused by loss of motor neurons in the spinal cord, brainstem and motor cortex, and can occur at any time in adulthood. Median survival is 3 years after symptom onset and is modestly prolonged by riluzole. A more effective treatment is urgently needed.
Protein aggregates or ubiquitin positive inclusions, containing proteins that are mutated in small subsets of ALS patients, have been recognised as the pathological hallmark of ALS. As the removal of misfolded or damaged proteins is critical for optimal cell functioning, impairment of protein turnover may play an important role in the pathogenesis of ALS and other neurodegenerative disorders.1–3
Lithium is well known for its mood stabilising effects in bipolar disorders but is being increasingly recognised as a neuroprotective agent.4–6 An important pathway in exerting this neuroprotective effect is the promotion of protein clearance or autophagy.7 In an ALS SOD1 mouse model of ALS, lithium showed reduced α-synuclein, ubiquitin and SOD1 aggregates in motor neurons, and apart from its effects on protein clearance, lithium was found to increase the number of mitochondria and suppressed reactive astrogliosis.8
These promising results from preclinical studies led to the testing of lithium in patients, putting it forward as a disease modifying agent for ALS. In a relatively small pilot study of 44 patients, 16 of whom received lithium at a dose leading to plasma levels ranging from 0.4 to 0.8 mEq/l, a significant effect on survival was found at 12 and 15 months of follow-up along with a slowing of disease progression.8 Subsequently, a trial designed to detect a large effect, as found in the pilot study (40% decrease in the rate of functional decline), had to be stopped (for ineffectuality) at the first interim analysis at 6 months.9 A more modest effect of lithium or an effect occurring after prolonged treatment could not be ruled out.10 Also, no effect could be detected in trials comparing a low (subtherapeutic) and therapeutic doses of lithium,11 comparing the effect of lithium treated patients with historical controls12 or using self-reported patient data.13 However, a placebo controlled, randomised trial designed to detect an effect on survival was still considered necessary as has been repeatedly pointed out14 15 (for a survey of previous studies, see supplementary table 1, available online only).
The progressive and fatal nature of ALS increases the importance of minimising the burden for patients of participating in clinical trials. In addition, treatment of patients should not be delayed if a novel therapy is effective, but if treatment is ineffective, unnecessary continuation of a trial should be avoided. The sequential trial design requires, on average, fewer patients than traditional trial designs of equal power and permits discontinuation of a study as soon as enough evidence for a treatment effect or lack thereof is obtained.16–21
The objective of this study was to examine the effect of lithium compared with placebo as an add-on treatment together with riluzole on survival in ALS, performing a randomised, clinical trial with sequential analysis.
Methods
Study design
This was a sequential, randomised (stratified and balanced 1:1 randomisation), double blind, placebo controlled, parallel group trial conducted in The Netherlands (three sites) (phase IIb trial). This trial was performed according to the Medical Research Involving Human Subjects Act (WMO) and the International Conference on Harmonisation Good Clinical Practice (GCP) guidelines.
Patients
Patients were eligible if they were diagnosed as having clinically probable laboratory supported, probable or definite ALS according to the World Federation of Neurology El Escorial criteria.22 Other inclusion criteria were: use of riluzole (50 mg, twice daily); onset of symptoms at least 6 months and no longer than 36 months prior to inclusion; a forced vital capacity (FVC) of at least 70% of the predicted value based on gender, height and age (in the sitting position); age between 18 and 85 years; and written informed consent. Patients were excluded if they met any of the following criteria: tracheostomal ventilation of any type, non-invasive ventilation for more than 16 h/day or supplementary oxygen during the last 3 months prior to inclusion; any medical condition or intoxication known to have an association with motor neuron dysfunction which might confound or obscure the diagnosis of ALS; presence of any concomitant life-threatening disease or any disease or impairment likely to interfere with functional assessment; contraindications to lithium therapy (eg, confirmed renal insufficiency or cardiac arrhythmias); and other medication interacting with lithium with a significant risk of complications (eg, verapamil). All patients gave written informed consent, and the ethics committees of the participating institutions approved the study.
Study settings
This study took place at the national referral centres for patients with motor neuron disease in The Netherlands (Netherlands ALS Centre, a collaboration of the Departments of Neurology of the University Medical Centre Utrecht, Academic Medical Centre Amsterdam and Radboud University Medical Centre Nijmegen), from November 2008 to June 2011. An electronic case report form (eCRF) was used for optimal and timely communication between patients and investigators as well as across centres.
An external trial monitor was engaged to protect the rights and well being of trial participants, to verify the accuracy of the reported trial data and to guarantee that the conduct of the trial was in compliance with the currently approved protocol/amendments, with GCP guidelines, and with the applicable regulatory requirements. Internal audits with ‘source data verification’ were performed to provide this additional level of security.
Intervention
Patients were assessed for eligibility at the screening visit, including blood tests (leucocytes, sodium, potassium, calcium, creatinine, glucose and thyroid stimulation hormone), urine tests (albumin, sediment analysis and specific gravity), body weight and ECG (in patients over 60 years of age). After confirmation of eligibility, patients were randomly assigned to receive either lithium carbonate once daily orally (lithium carbonate 400 mg; PCH, Haarlem, The Netherlands) at a target concentration of 0.4–0.8 mEq/l or identical placebo tablets (manufacturing pharmacy: ‘Apotheek Haagse Ziekenhuizen’, The Hague, The Netherlands). Both lithium carbonate and placebo tablets were packed in identically labelled containers by ‘Apotheek Haagse Ziekenhuizen’. The manufacturing pharmacy was not involved in the study design and they did not provide material support. Trial medication was handed out to the patients or sent by courier by the pharmacy of UMC Utrecht.
Patients were instructed to commence with one tablet in the evening, and a first control of lithium concentration took place after 1 week. To minimise the burden for patients, patients were bled at the general practitioner's surgery or local laboratory in return for an allowance (samples were obtained approximately 12 h after the last dose). Samples were sent by mail to a central laboratory (at UMC Utrecht) where blood concentrations were measured and the result was entered into the eCRF. For reasons of safety monitoring, patients completed a questionnaire in the eCRF reporting on possible side effects: polydipsia, polyuria, vomiting, diarrhoea, fatigue, weight gain, tremor, concentration and/or memory problems, movement disturbances, skin changes and miscellaneous. An unblinded investigator (JHV) received an instant notification by email to make dose adjustments. Then, the patient and local investigator received an email about the new dosage and timing of the next blood control. As soon as the lithium concentrations were within the target range (0.4–0.8 mEq/l), the control frequency was lowered to a minimum of once every 3 months. Patients who received placebo were given simulated dose modifications identical to the lithium group.
Outcomes
The primary outcome measure with respect to efficacy in ALS was survival, defined as the time from inclusion to death, tracheostomal ventilation or non-invasive ventilation for more than 16 h/day. Caregivers and/or patients were instructed to report endpoints immediately. We chose survival as the primary outcome measure as a relevant and sensitive means of determining a potential disease modifying effect.23
The rate of decline in daily functioning, measured by the validated revised ALS Functional Rating Scale (ALSFRS-R) and FVC were used as secondary outcome measures.24 Unintended effects of the intervention were assessed by registration of all (serious) adverse events.
Secondary outcome measures were assessed during the visits every 3 months. In addition, body weight was assessed and blood samples were taken to measure lithium concentration, thyroid function (thyroid stimulating hormone), kidney function (creatinine), electrolytes (sodium, potassium), liver enzymes (aminotransferase concentrations, alkaline phosphatase, γ-glutamic transpeptidase) and leucocytes (at the first two visits). Patients who were not able to visit the hospital because of disease progression were called by telephone to document (serious) adverse events and ALSFRS-R. Serious adverse events (SAEs), as defined in the GCP guidelines, were documented in the eCRF from which an automatic notification email was generated to the coordinating investigator. The ethics committee was notified within 2 weeks about SAEs judged to be related to therapy and received a safety report of all adverse events (SAEs and AEs) twice a year. As of 1 January 2010, the ethics committee was notified electronically about all serious adverse reactions and events within 15 days and in case of a lethal or life-threatening SAE, within 7 days.
Sample size
The trial was designed to detect a 15% increase in cumulative survival percentage in the lithium group, similar to the effect of riluzole.25 Assuming a cumulative survival percentage in the placebo group of 60% after 16 months and 75% in the lithium group (ie, a HR of 0.56 for lithium compared with placebo), with a one sided α level of 0.05 and a power (1−β) of 90%, on average 173 patients with 56 endpoints (90th percentile=283 patients) would be required if the null hypothesis were true (ie, no difference between placebo and lithium), and on average 191 patients with 62 endpoints (90th percentile=311 patients) if the alternative hypothesis were true (treatment with lithium is superior to placebo).
This sample size calculation was based on the sequential trial design which allows the study to be halted as soon as the cumulative data show the efficacy of the studied drug or a lack thereof.21 This implies that the total number of patients to be included cannot be estimated beforehand but is determined by the course of the trial. For comparison, the total fixed sample size for a classic phase IIb randomised controlled trial, based on the same expected difference in survival of 15% with a one sided α of 0.05 and a power of 0.90, would be at least 338 patients with approximately 110 endpoints.
Interim analyses and stopping guidelines
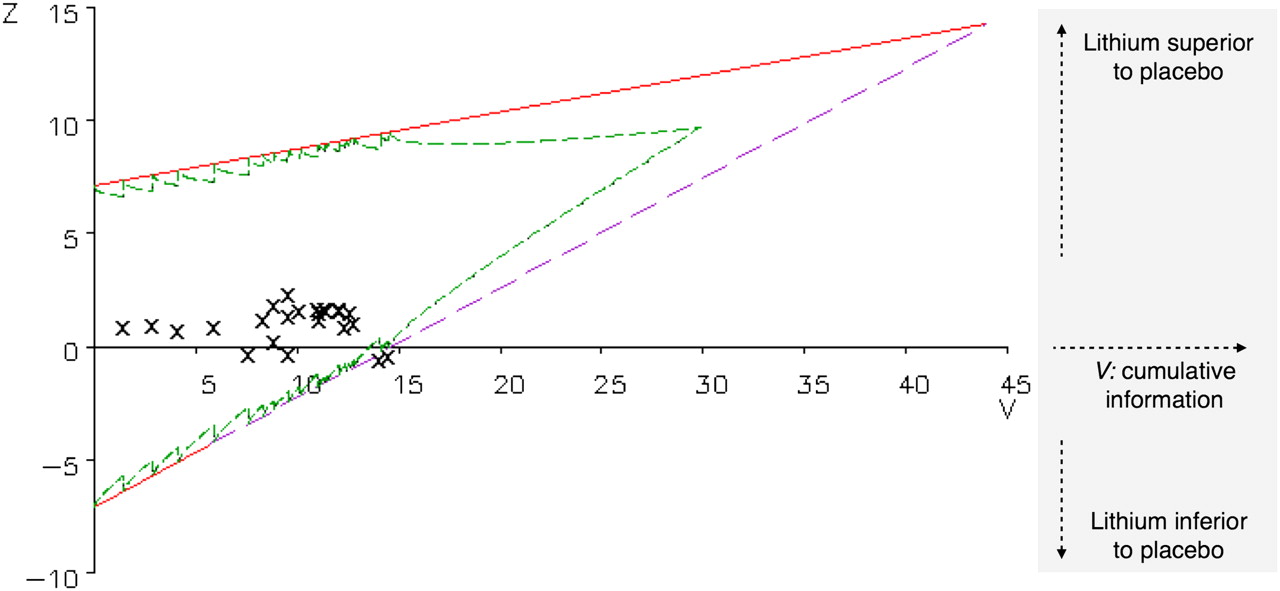
A sequential design for an randomised controlled trial allows a series of interim analyses on the emerging data to be conducted and specifies the circumstances that determine when the trial will stop or continue at each analysis. Sequential monitoring of the emerging survival data was conducted by an independent biostatistician (IvdT). Based on a 15% difference in cumulative survival, one sided type I error of 0.05 and power of 90%, monitoring boundaries were specified. These boundaries are determined so that the probability of falsely declaring an ineffective treatment as effective remains controlled at 5% and the probability of incorrectly declaring an effective treatment to be ineffective remains controlled at 10%, wherever the boundaries are crossed. These monitoring boundaries are straight lines, based on an idealised version of continuous monitoring (see figure 1 for illustration). The analysis is not truly continuous but is performed on each new set of cumulative data, based on the occurrence of one or several new clinical endpoints. This (discrete) approximation of a truly continuous analysis is graphically depicted by the jagged inner lines below the upper and above the lower limit (the so-called ‘Christmas tree correction’).21 A statistic Z is defined to measure the treatment effect as the difference between the observed number of events in the control group and the expected number under the assumption of treatment equivalence. A positive Z value indicates that the treatment is superior; a negative value indicates that the treatment is equal or inferior to placebo. Each new clinical endpoint or group of clinical endpoints leads to a new value for Z. The cumulative amount of information (concerning events) is expressed in the statistic V. For a sequential survival analysis, V is approximately equal to a quarter of the number of events observed. When the test statistic based on the cumulative data crosses the upper boundary, the null hypothesis of indifference is rejected. When the lower boundary is crossed, the null hypothesis is accepted (figure 1). The effect size of lithium relative to placebo in terms of the HR was specified at 16 months to be able to calculate the monitoring boundaries and to determine the average number of patients required, thus assuming a constant proportional HR independent of follow-up time. It is nevertheless possible to continue monitoring patients after 16 months of follow-up. Data on patients reaching an endpoint after 16-months can therefore be included in the sequential survival analysis.

Flow diagram.
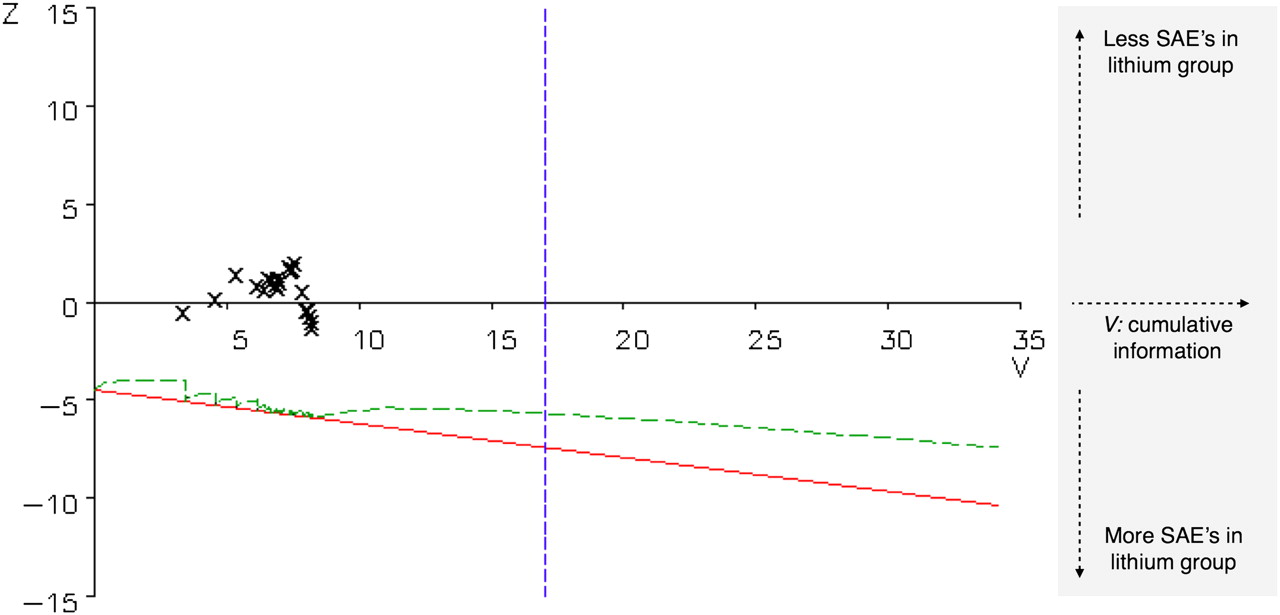
In addition to the efficacy analysis, a safety analysis was performed comparing the proportion of patients who had a serious adverse event in the lithium group with those in the placebo group. Safety was also monitored sequentially.21 26 The safety boundary is determined by the hypothesised SAE rates for the lithium and placebo group as well as the risk of false alarm α. An SAE rate of 0.25 was expected in the placebo group; an SAE rate of 0.40 or more in the lithium group was considered undesirable. The type I error α was set at 0.10 (one sided) as a warning when lithium would be unsafe. An independent data and safety monitoring board (DSMB) was installed, composed of people acquainted with either lithium, clinical trials and/or ALS (eg, clinical pharmacologist, psychiatrist, nephrologist, trial neurologist and a biostatistician). The DSMB periodically reviewed the efficacy and safety data.
Randomisation and blinding
Stratified randomisation to receive lithium or placebo was performed manually by one of the investigators not involved in the care of trial patients (JHV). To ensure a balanced number of patients receiving lithium and placebo in the strata, the minimisation method, as described by Pocock,27 was applied stratified to the following prognostic factors: age (<45; ≥45 and ≤65 years; or >65 years), site of symptom onset (bulbar or spinal), FVC (<85% or ≥85%) and location (University Medical Centre Utrecht, Academic Medical Centre Amsterdam or Radboud University Medical Centre Nijmegen). The patient was assigned a unique medication code corresponding to either placebo or lithium. As such, unblinding of a single participant would not result in unblinding of the whole group. Besides the randomising investigator, only the pharmacy and the biostatistician had access to the randomisation sheet until the end of the trial. As each of the containers with trial medication was provided with a unique medication code, additional dispenses of trial medication to the same patient had a different code but (obviously) corresponded to the same treatment arm.
Patients were assessed for eligibility at the screening visit. After confirmation of eligibility, the local investigator asked the randomising investigator by email (including study number of the patient and the essential information for minimisation) to randomise the patient. The medication code assigned to the patient was entered on the eCRF.
Statistical methods
The sequential design and analysis has been described above (‘Interim analyses and stopping guidelines’). Sequential analysis was stratified for the factors described (‘Randomisation and blinding’). For descriptive reasons, Kaplan–Meier survival curves were estimated. The estimate for the HR has to be adjusted for the fact that the data are analysed sequentially. PEST 4 software was used for the sequential analysis.16 26
To estimate the difference in rate of decline in functional status and respiratory function, measured by the ALSFRS-R and FVC, we fitted the longitudinal data by maximum likelihood estimation using linear mixed effects models, including time as a continuous variable and treatment group as a factor, adjusting for gender and the stratification factors as categorical variables (age, FVC, site of onset and centre). A possible overall non-linear decline of the ALSFRS-R and FVC could be accounted for by also running the model with time as a categorical variable (visit number) because slopes between visit numbers are compared between treatment groups. In addition, we explored additional models with time entered as an exponential continuous variable (powers 2, 4 and the square root). SAEs were monitored as described above (‘Interim analyses and stopping guidelines’). All adverse events were categorised per organ system and specific event, and compared between groups using Fisher's exact tests. All results were analysed on an intention to treat basis.
Results
Patients
From November 2008 until June 2011, 155 patients were considered for enrolment. Twenty-two patients did not fulfil the inclusion criteria (figure 1). After screening, 133 patients were randomly assigned to receive lithium (n=66) or placebo (n=67). Baseline characteristics of the participants are shown in table 1. Thirty patients (45%) from the lithium group and 20 patients (30%) from the placebo group discontinued trial medication after on average of 10 months of treatment. Significantly more patients in the lithium group discontinued trial medication due to one or more side effects (p=0.009; fatigue or a general feeling of discomfort (lithium group: n=5; placebo group: n =1), tremor (lithium group: n=5), psychological complaints (lithium group: n=3; placebo group: n=2), polyuria/nocturia (lithium group: n=3; placebo group: n=1), nausea (lithium group: n=3), skin changes (lithium group: n=1; placebo group: n=2), elevated liver enzymes (lithium group: n=1), restless legs (lithium group: n=1), headache (placebo group: n=1) and palpitations (placebo group: n=1)) (figure 1). All randomised patients were included in the final analysis on the primary outcome. One participant was lost to follow-up for the secondary outcome measures shortly after randomisation (no post-randomisation measures were obtained); therefore, this patient was not included in these analyses. Median follow-up was 16 months for the lithium group (n=66) and 15 months for the placebo group (n=67) (IQR 2–31 months for both groups).
Baseline characteristics of the study participants
Primary outcome measure: survival
After 61 patients had reached a primary endpoint (lithium group: n=33; placebo group: n=28), the sample path of (Z, V) values (the X symbols in figure 2) crossed the lower boundary, indicating that the null hypothesis of no difference between the two treatment arms could not be rejected (p=0.91; figure 2). The cumulative survival probability at 12 months was 0.73 in the lithium group (SE 0.06; 95% CI 0.63 to 0.86) versus 0.75 (SE 0.06; 95% CI 0.65 to 0.87) in the placebo group, and at 16 months 0.62 in the lithium group (SE 0.06; 95% CI 0.50 to 0.76) versus 0.67 (SE 0.06; 95% CI 0.56 to 0.81) in the placebo group (figure 3). The adjusted HR was 1.03 for lithium compared with placebo, with 90% CI 0.66 to 1.63. Of the 61 patients who reached the primary endpoint, 51 patients died, nine patients were on non-invasive ventilation for more than 16 h/day and in one patient tracheostomal ventilation was initiated (figure 1).

Sequential survival analysis. On the horizontal axis, the amount of information V increases as time passes and events accumulate. On the vertical axis, Z (cursive) reflects the effect of lithium relative to placebo. When the sample path depicted by the X symbols crosses the upper (inner) boundary, the null hypothesis (ie, no treatment effect) is rejected: the effect size is significant. When the test statistic crosses the lower (inner) boundary, it becomes highly improbable that the upper boundary of the triangle will also be crossed, and the null hypothesis of indifference cannot be rejected. Crossing of the first (red) part of the lower boundary indicates a negative effect of lithium on survival. The (green) inner margins of the triangle are the true boundaries to be crossed (the so-called ‘Christmas tree correction’) whereas the straight lines that form the upper (red) and lower (broken purple) boundary are the hypothetical boundaries to be crossed if the analysis were truly sequential.

Kaplan–Meier survival curve. Cumulative survival for patients treated with lithium compared with the placebo group.
A post hoc on-treatment analysis (patients on lithium for <3 months were considered as placebo, n=9) using a log rank test was found to be similar to the findings in the intention to treat analysis (p=0.50).
Secondary outcome measures: daily functioning and vital capacity
Figure 4 shows the decline in mean ALSFRS-R and FVC for both treatment groups during the course of the trial. Linear mixed effects modelling—adjusted for gender and stratification factors—showed that the interaction between treatment group and time was not statistically significant for ALSFRS-R (p=0.74) and FVC (p=0.22). Similar results were obtained by including time as a categorical variable (visit number) or time as an exponential continuous variable in the model, accounting for a possible overall non-linear decline in ALSFRS-R and FVC.

Secondary outcome measures. The level of daily functioning measured by the revised Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS-R) and forced vital capacity (FVC) for the lithium group compared with the placebo group. The decline over time does not differ significantly between treatment groups. As FVC measures could not be obtained in advanced stages of the disease, follow-up time is shorter compared with ALSFRS-R measures. It is important to note that the number of patients included in these analyses is declining over time as patients are sequentially included and die during the course of their disease (numbers at risk are given in figure 3).
Lithium blood concentrations
Target blood concentrations (0.4–0.8 mEq/l) were established in 73% of patients on lithium. In addition, 87% of the measures were between 0.3 and 1.0 mEq/l. In a few patients, consistent dose dependent side effects resulted in subtarget blood concentrations.
Adverse events
In the placebo group 35 of 67 patients and in the lithium group 39 of 66 patients experienced one or more SAEs, which was not significantly different in the sequential safety monitoring, as shown in figure 5. Comparison of specific SAEs also showed no relevant differences (table 2). One patient was unblinded after a toxic lithium concentration (1.9 mEq/l) was measured. This patient suffered from rapid progression of bulbar weakness resulting in severe weight loss and dehydration. Study medication was discontinued but despite treatment with infusion of fluids and gastric tube placement she died shortly after this episode due to progressive respiratory failure. Another six SAEs in five patients were reported as possibly being related to the study medication by the (blinded) site investigators (chest pain/palpitations: n=3; restlessness/confusion: n=2; dehydration: n=1). Three of these five patients received lithium.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Sequential safety monitoring. On the horizontal axis, the amount of information V increases as time passes and serious adverse events (SAEs) accumulate. On the vertical axis, Z reflects the effect of lithium relative to placebo. If the sample path depicted by the X symbols were to cross the lower boundary, there would possibly be a safety problem for lithium. The (green) inner boundary is the true boundary to be crossed (the so-called ‘Christmas tree correction’) whereas the straight line that forms the lower (red) boundary is the hypothetical boundary to be crossed if the analysis were truly sequential. The vertical blue boundary indicates the V value corresponding to a sample size of 311.
Overview of serious adverse events
Comparison of the classified adverse events—as reported by either investigators or patients—showed several events occurring in significantly more patients on lithium treatment: nausea (lithium group: n=17; placebo group: n=6; p=0.011), vomiting (lithium group: n=12; placebo group: n=2; p=0.0043) and polydipsia (lithium group: n=37; placebo group: n=23; p=0.014). In supplementary table 2 (available online only), the AEs are listed per organ system.
Discussion
This study showed that lithium, in combination with riluzole, did not improve survival in patients with ALS. Lack of efficacy was established after the lower boundary of the sequential analysis was crossed, ruling out a modest beneficial effect of lithium, defined as a 15% increase in cumulative survival percentage. Secondary outcome measures supported the equivalence of lithium and placebo based on disease progression, as measured by the ALSFRS-R and vital capacity. Safety analyses did not reveal major safety issues but discontinuation of trial medication due to adverse effects occurred significantly more often in patients taking lithium compared with placebo.
Following the first pilot study,8 preclinical studies with lithium could not consistently replicate the initial findings.28 29 In addition, a web based observational study and five controlled clinical studies were initiated. It is noteworthy that the clinical trials with lithium published thus far have not focused solely on studying the efficacy of lithium but have also explored new trial designs9 11–13 (see supplementary table 1, available online only). The only placebo controlled trial thus far has efficiently ruled out a large and early effect of lithium.9 However, we now report on a placebo controlled trial ruling out a modest or delayed effect of lithium on cumulative survival using a sequential trial design.16 18 20 This design saved ‘time and patients’ as due to the sequential analysis, the trial was stopped as soon as there was enough evidence for a lack of treatment effect. In addition, the inefficacy of lithium for the treatment of ALS was demonstrated despite the inclusion of fewer patients than the calculated sample size had it been a fixed size trial design.
Defective cellular protein clearance (or autophagy) is increasingly recognised as an important mechanism of neurodegeneration in ALS.1–3 These insights draw attention to candidate drugs promoting protein clearance, such as lithium. A well conducted, randomised, clinical trial which provides a definite answer regarding the efficacy of lithium is mandatory. The current trial does not lend support to a beneficial effect of lithium on survival in ALS.
An important aim of this study was to assess the safety of lithium for the treatment of patients with ALS. The use of online questionnaires assessing potential side effects yielded a large volume of safety data. Indeed, well known side effects of lithium—for example, nausea and polydipsia—occurred more frequently in the lithium treated group. In line with a previous report, significantly more patients in the lithium group experienced these (and other) side effects which led to discontinuation of trial medication.11 Fortunately, we did not find any significant differences regarding the occurrence of SAEs. Therefore, we conclude that despite the known inconvenient side effects, lithium can be safely given to patients with ALS.
The population participating in trials has been observed to be different from the incident cohort of patients with ALS. These differences are partly due to the inclusion and exclusion criteria, as adopted in clinical trials, and partly to the type of patients choosing to participate.30 However, as most patients in this sequential trial were included shortly after diagnosis, the clinical characteristics approached the incident cohort. This is best reflected by the 29% bulbar onset patients participating, which is close to the 30% bulbar onset based on epidemiological data.31 A second limitation is the lack of different dosages as it may be hypothesised that the dosage used was not sufficient to induce effects. We examined the same dosage used in the initial pilot study, as this was found to be effective. A previous report comparing subtherapeutic (0.2–0.4 mEq/l) with therapeutic lithium treatment found no dose dependent effect.11 None of the previous studies on lithium in ALS have included higher dosages. As for the narrow therapeutic range of lithium and therefore the increased risk of toxicity, we refrained from including a higher dosage group out of concern for the safety of our patients. Third, the dropout rate for lithium trials tends to be higher compared with trials with other compounds in ALS, which might have obscured the observed effects. We therefore performed an on-treatment analysis confirming the lack of efficacy similar to the findings in the intention to treat analysis. Finally, it is noteworthy that little is known about potential interactions between lithium and riluzole. However, the limited evidence available suggests a potentiating rather than a detrimental effect of riluzole on the neuroprotective properties of lithium.32
In conclusion, this randomised, sequential, placebo controlled trial demonstrated the inefficacy of lithium for the treatment of ALS based on cumulative survival in a substantial cohort of patients. The use of a sequential trial design precludes unnecessary delay if the study drug is effective as well as undesirable continuation of an ineffective or even harmful treatment. The current results provide physicians with the evidence needed to adequately advise their patients with ALS on the use of lithium.
Acknowledgments
The authors are grateful to the patients and caregivers for their efforts and time. The authors thank the members of the data and safety monitoring board for their guidance, and the devoted trial nurses Inge van Beilen, Dorien Standaar, Martha Huvenaars and Lotte Knapen.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
Funding The authors are grateful for funding of this study from the ‘Zeldzame Ziekten Fonds’, The Zabawas Foundation, The Netherlands ALS Foundation, Optimix Foundation and Jan Cornelia Foundation.
Competing interests None.
Ethics approval The study was approved by the Medical Ethics Committee for Research into Humans of the University Medical Centre Utrecht (and received local approval from the Medical Ethics Committee for Research into Humans of the Academic Medical Centre, Amsterdam and Radboud University Medical Centre, Nijmegen).
Provenance and peer review Not commissioned; externally peer reviewed.