Article Text

Abstract
Background Charcot–Marie–Tooth disease (CMT) is a clinically and genetically heterogeneous group of diseases with approximately 45 different causative genes described. The aims of this study were to determine the frequency of different genes in a large cohort of patients with CMT and devise guidelines for genetic testing in practice.
Methods The genes known to cause CMT were sequenced in 1607 patients with CMT (425 patients attending an inherited neuropathy clinic and 1182 patients whose DNA was sent to the authors for genetic testing) to determine the proportion of different subtypes in a UK population.
Results A molecular diagnosis was achieved in 62.6% of patients with CMT attending the inherited neuropathy clinic; in 80.4% of patients with CMT1 (demyelinating CMT) and in 25.2% of those with CMT2 (axonal CMT). Mutations or rearrangements in PMP22, GJB1, MPZ and MFN2 accounted for over 90% of the molecular diagnoses while mutations in all other genes tested were rare.
Conclusion Four commonly available genes account for over 90% of all CMT molecular diagnoses; a diagnostic algorithm is proposed based on these results for use in clinical practice. Any patient with CMT without a mutation in these four genes or with an unusual phenotype should be considered for referral for an expert opinion to maximise the chance of reaching a molecular diagnosis.
Statistics from Altmetric.com
Introduction
The inherited neuropathies are a genetically and clinically heterogeneous group of disorders encompassing Charcot–Marie–Tooth disease (CMT), hereditary neuropathy with liability to pressure palsy (HNPP), hereditary motor neuropathy (HMN) and hereditary sensory and autonomic neuropathy (HSAN, also known as hereditary sensory neuropathy). Mutations in over 45 distinct genes have been implicated in causing the inherited neuropathies, and many more remain unknown.1 CMT is the most common inherited neuromuscular disorder, affecting 1 in 2500.2 Inheritance may be autosomal dominant, autosomal recessive or X linked; however, de novo dominant cases occur relatively frequently in CMT and thus some patients may not have a family history of CMT. Neurophysiology differentiates CMT into demyelinating CMT1 (upper limb motor nerve conduction velocity (MNCV) <38 m/s), axonal CMT2 (MNCV >38 m/s) and intermediate CMT (ICMT, MNCV 25–45 m/s). CMT1 occurs more frequently than CMT2; this may be partially explained by the fact that the majority of causative genes for CMT2 remain undetermined.3 4
The classical CMT phenotype is of onset within the first two decades with difficulty walking, sensory loss, foot deformities and signs of a length dependent sensorimotor neuropathy. However, some patients present early with much more severe disease while others remain asymptomatic until adulthood. To some extent the underlying genetic cause explains the variable phenotypes seen in CMT although there is significant overlap; mutations in many different genes cause a similar phenotype and, conversely, mutations in the same gene may cause different phenotypes.1 This clinical and genetic heterogeneity makes diagnosis and genetic counselling difficult for clinicians. A molecular diagnosis is also useful in order to guide prognosis. More importantly, now that clinical trials of treatment have commenced for some types of CMT (eg, ascorbic acid for CMT1A),5 an accurate genetic diagnosis is essential.
As part of our peripheral nerve service in the National Hospital for Neurology and Neurosurgery, we run an inherited neuropathy clinic. We also run a diagnostic laboratory for some of the common CMT genes (chromosome 17 dosage analysis, sequencing of PMP22, GJB1, MFN2, MPZ, GDAP1, BSCL2 and SPTLC1) and, in addition, are sent many DNA samples from patients with CMT for research testing of the less common CMT genes from throughout the UK. As a result, we are well placed to determine the frequency of the different genetic subtypes of CMT in our patient cohort and devise a strategy to guide genetic testing for clinicians.
Patients and methods
This study was approved by the research ethics committee of the National Hospital for Neurology and Neurosurgery. All patients gave written informed consent to undergo genetic testing.
Patient cohort
Since 2006, a detailed database has been kept documenting all patients seen in our inherited neuropathy clinic. In addition, the database includes details of external patients whose DNA samples were sent to us for diagnostic and/or research testing. We included isolated patients as well as those with a family history. Details recorded include patient age, type of inherited neuropathy (CMT1, CMT2, ICMT, HNPP, HMN, HSAN), family history and other clinical features, including neurophysiology and the CMT neuropathy score (CMTNS)6 or the CMTNS2,7 where available.
For patients seen in our inherited neuropathy clinic, detailed information regarding phenotype was available. Clinical diagnosis was based on symptoms, signs, family history (including assessment of family members when possible) and neurophysiology, and patients were classified into subgroups (eg, CMT1, CMT2, ICMT) as previously defined by us.8 9 For external patients, more limited information was available and diagnosis was based on the information received. In order to distinguish CMT from HMN, both clinical and neurophysiological evidence of sensory involvement was taken into account. Patients with a predominantly sensory neuropathy (sensory axonal neuropathy characterised by loss of sensation, including pain and temperature with or without positive sensory symptoms, such as pain and paraesthesiae and ulceromutilating complications) were classified as HSAN.10 Patients with complex neurological diseases who had neuropathy as part of the phenotype (eg, ataxia with oculomotor apraxia) were classified as having a complex neuropathy. The molecular part of this study concerns those patients we classified as having a primary inherited neuropathy (CMT, HNPP, HMN and HSAN) with a more detailed analysis of those patients classified as having CMT. We excluded patients with a complex neuropathy from the molecular study.
We determined the frequency of each subtype of CMT based on the number of patients with a particular subtype out of the total number of index patients. As we perform genetic testing sequentially, all patients have not had all possible genes tested and this should be kept in mind when interpreting the results. We also determined ‘hit rates’ for mutations in specific rare genes by calculating the number of positive results found out of the total number of patients for whom testing was performed.
Molecular genetic analysis
For this study, as well as the genes screened in our diagnostic laboratory (chromosome 17 dosage analysis, PMP22, GJB1, MFN2, MPZ, GDAP1, BSCL2 and SPTLC1) we also screened LITAF, SH3TC2, MTMR2, EGR2, NEFL, TRPV4, HSPB1, HSPB8 and GAN1 where appropriate. LITAF and SH3TC2 were screened in patients with CMT1 who were negative for PMP22 rearrangements; TRPV4, HSPB1 and HSPB8 were screened in patients with CMT2 who were negative for mutations in MFN2 as well as in patients with HMN; MTMR2 was screened in a select group of patients with early onset severe autosomal recessive CMT1 negative for PMP22 rearrangements; and EGR2 and NEFL were screened in patients with CMT1 negative for rearrangements of PMP22 or in patients with CMT2 negative for mutations in MFN2.
Dosage analysis of the 17p region, including PMP22, was performed by semiquantitative fluorescent PCR or multiplex ligation dependent probe amplification. For sequencing, coding exons and flanking intronic regions were amplified using primer oligonucleotides and Roche (Mannheim, Germany), Applied Biosystems (Foster City, California, USA) or Qiagen (Hilden, Germany) PCR kits, according to the manufacturers' instructions. Primers and PCR conditions are available on request. Sequence reactions were performed using Big Dye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems), and products were cleaned using Dye Terminator removal plates (Abgene, Vilnius, Lithuania). Sequencing products were resolved on an AB 3730xl Sequencer. The resulting sequences were aligned and analysed with SeqScape (Applied Biosystems) or Sequencher (Gene Codes Corporation) software. Variations were confirmed by repeat sequencing. Segregation of mutations within families was performed where possible. Where novel variations were found, control groups of UK or Asian chromosomes were screened according to the ethnicity of the patient. Mutations were considered potentially pathogenic if they were absent from controls and segregated with disease in the family. We also considered conservation of amino acids among species and used three commonly used prediction programs, PolyPhen (http://genetics.bwh.harvard.edu/pph/), SIFT (http://blocks.fhcrc.org/sift/SIFT.html) and aGVGD (http://agvgd.iarc.fr/) to help decide on the pathogenicity of the mutations.
Results
A total of 2732 patients are included in our neuropathy database, 2094 of whom have a diagnosis of a primary inherited neuropathy (76.6%) (CMT, HNPP, HMN, HSAN). The remaining 638 individuals include unaffected family members and patients with more complex neurological conditions (complex neuropathy) who have neuropathy as part of their syndrome (eg, autosomal recessive spastic ataxia of Charlevoix–Saguenay, ataxia with oculomotor apraxia, etc). A total of 916/2732 (33.5%) patients were seen in our inherited neuropathy clinic between 2006 and 2011 and more detailed phenotype information is available for this subgroup.
Inherited neuropathy clinic cohort
Of the 916 patients seen in our inherited neuropathy clinic, 601 (65.6%) had a primary inherited neuropathy (425 CMT, 46 HNPP, 61 HMN, 69 HSAN). Of the 425 patients with CMT, 240 had CMT1 (56.5%), 115 had CMT2 (27.1%), 62 had ICMT (15.6%) and eight (1.9%) were unclassified (usually because the patient refused nerve conduction studies or had unrecordable responses on nerve conduction studies).
Overall, 266/425 (62.6%) patients with CMT received a molecular diagnosis: 193/240 (80.4%) patients with CMT1, 29/115 (25.2%) patients with CMT2 and 37/62 (59.7%) patients with ICMT (see table 1 for the breakdown of genetic diagnoses). Two hundred and forty-five of the 266 molecular diagnoses (92%) were accounted for by mutations or rearrangements of PMP22, GJB1, MFN2 or MPZ.
Molecular diagnoses in patients with Charcot–Marie–Tooth disease attending an inherited neuropathy clinic
Of the 46 patients with a clinical diagnosis of HNPP, 27 (58.7%) had the PMP22 deletion and four (8.7%) had a point mutation in PMP22. Of the 61 patients with HMN, 10 (16.4%) had a molecular diagnosis; five HSPB1, two SMN1, one GARS, one HSPB8 and one BSCL2. Of the 69 patients with HSAN, 14 (20.3%) had mutations in SPTLC1, five (7.2%) SPTLC2, one (1.4%) heterozygous NGFB and one FAM134B (1.4%).10
The CMT subtype associated with each gene was in keeping with published data (table 2). Analysing the common genes, patients with the PMP22 duplication all had CMT1, those with MFN2 mutations all had CMT2 and patients with GJB1 or MPZ mutations had a range of phenotypes, including CMT1, CMT2 or ICMT. The phenotypes of the rarer forms of CMT are summarised in table 2.
Charcot–Marie–Tooth disease subtype associated with individual genes in patients attending an inherited neuropathy clinic
Patients not attending inherited neuropathy clinic
Of the 1816 patients not attending the inherited neuropathy clinic, 1493 (82.2%) had a primary inherited neuropathy: 1182 (65.1%) CMT, 56 (3.1%) HNPP, 126 (6.9%) HMN and 129 (7.1%) HSAN.
Of the 1182 patients with CMT, 446 had CMT1 (37.7%), 335 had CMT2 (28.3%), 23 had ICMT (0.4%) and 378 were unclassified (32%) (type of CMT not documented on genetic request form). Overall, 446/1182 (37.7%) patients with CMT received a genetic diagnosis: 269/446 (60.3%) with CMT1, 44/335 (13.1%) with CMT2, 5/23 (21.7%) with ICMT and 128/378 (33.9%) of the unclassified group. Four hundred and nineteen of the 446 patients who achieved a molecular diagnosis (94%) were accounted for by mutations or rearrangements of PMP22, GJB1, MFN2 or MPZ (table 3).
Genetic diagnoses in patients with Charcot–Marie–Tooth disease not attending an inherited neuropathy clinic
When the molecular diagnosis rate was compared in patients attending the inherited neuropathy clinic with those that did not attend our clinic, the diagnosis rates were significantly different for all groups (table 4). Overall, a molecular diagnosis was achieved in 62.6% of patients attending the inherited neuropathy clinic versus 37.7% in those not attending this clinic (p=0.003).
Molecular diagnosis rate
Hit rates for specific genes
We determined hit rates for the genes not available in the diagnostic laboratory. Mutations in these genes are thought to be rare which is why a diagnostic test is not widely available. Large numbers of patients (see table 5 for numbers) with CMT1 were screened for LITAF, EGR2 and SH3TC2; patients with CMT1 or CMT2 were screened for NEFL; and patients with CMT2 or HMN were screened for TRPV4, HSPB1 and HSPB8. A select group of patients with early onset severe CMT1 were screened for MTMR2. We found mutations in all genes tested (table 5); each (other than MTMR2 for which only nine patients were tested) accounted for <3% of patients who did not have mutations in the genes that most commonly cause CMT (PMP22 duplication in CMT1 and MFN2 in CMT2).
Hit rates for rare Charcot–Marie–Tooth genes
Discussion
In this study, we detailed the frequency of different primary inherited neuropathies (CMT, HNPP, HMN and HSAN) in both a cohort of patients attending a specialised inherited neuropathy clinic and in a large cohort of patients whose DNA was sent to us for molecular testing. Furthermore, we determined the molecular diagnosis rate in patients attending a specialist inherited neuropathy clinic and in those in the larger cohort, and also determined the hit rate for specific rare CMT genes.
We found that CMT1 was more common than CMT2 (56.5% vs 27.1%) in the inherited neuropathy cohort; this is similar to the distribution found in a recent epidemiological study from Northern England that found that 56.7% of their CMT cohort had CMT1 and 17.6% had CMT2.3 A Norwegian study found roughly equal distributions of CMT1 and CMT2 (48.2% vs 49.4%)11; however, Norway has a higher prevalence of CMT than other European countries3 12 and has a relatively isolated genetic population which might explain the differing results.
A molecular diagnosis was achieved in 62.6% of patients with CMT attending our inherited neuropathy clinic. Saporta et al performed a similar study in 2011 of 787 patients with CMT attending an inherited neuropathy clinic in Detroit where a molecular diagnosis was made in 67% of cases.4 This study included HNPP within the CMT cohort, which accounted for 6.1% of all of their CMT patients, thus the molecular diagnosis rate in CMT alone was 60.9%, very similar to our findings. The majority of molecular diagnoses in our study were accounted for by rearrangements or mutations in four genes: PMP22, GJB1, MFN2 and MPZ. These four genes accounted for 92% of the molecular diagnoses in patients attending our inherited neuropathy clinic and 94% of the molecular diagnoses in other patients. Similarly, the same four genes accounted for 91% of genetically determined CMT in the Detroit cohort.4
Greater than 80% of patients with CMT1 received a molecular diagnosis, mostly accounted for by the PMP22 duplication (70%). This is in keeping with other European estimates which found that 70.7% of CMT1 patients had the PMP22 duplication.13 In fact, the PMP22 duplication accounted for 39.5% of all CMT, similar to the 36.9% in Detroit4 and 42.8% in Northern England.3 In contrast, only 25.2% of patients with CMT2 received a molecular diagnosis, the majority caused by either MFN2 or GJB1 mutations. Although a lower proportion of CMT2 (10.4%) was accounted for by MFN2 mutations than suggested by other studies,14 overall, MFN2 mutations accounted for 2.8% of CMT, similar to that found by Saporta et al (2.7%). This may reflect a referral bias, as many patients with CMT2 referred to our clinic have had MFN2 excluded prior to referral. Given that most known CMT2 genes each account for a small proportion of CMT2 families, next generation sequencing technology is likely to lead to increasing numbers of genes for CMT2.15
All other genes tested accounted for less than 3% of CMT each in a large cohort of patients who were negative for either the PMP22 duplication (in patients with CMT1) or MFN2 mutations (in patients with CMT2). This suggests that mutations in these genes are rare causes of CMT in the general CMT population. This is in keeping with the literature which suggests that LITAF mutations are found in 0.6–3.75% of CMT1,16 17 SH3TC2 mutations in 21.7% of recessive CMT1 and 0.4% of all CMT,4 18 19 EGR2 mutations in 0.1–2% of CMT1,16 20 NEFL mutations in 0.5–3% of CMT116 21 and 2% of all CMT,21 HSPB1 mutations in 1.4–4.8% of CMT2/HMN22 23 and HSPB8 mutations in 0–2.6% of CMT2/HMN.23–25 Patients with CMT due to mutations in these rare genes often have specific phenotypic clues to the diagnosis; for example, CMT4C due to recessive mutations in SH3TC2 usually presents early with a demyelinating neuropathy and associated scoliosis and has characteristic features of elongated Schwann cell processes on nerve biopsy,19 thus the diagnosis may be more likely to be reached in a specialist inherited neuropathy clinic.
The molecular diagnosis rate was significantly lower in patients who were not seen in the inherited neuropathy clinic (37.7%). This is partly due to the high proportion of DNA samples sent for genetic testing without specifying the subtype of CMT (32%). The practice in our diagnostic laboratory is to screen the genetic test requested; that is, if a DNA sample is sent requesting PMP22 duplication, this test is performed even if the type of CMT is not specified. In this situation, we cannot ensure that the appropriate genetic test is performed, or give advice on further genetic testing. We receive some requests for PMP22 duplication testing in patients documented to have CMT2 although no patient with a PMP22 duplication has ever been documented to have an axonal phenotype; thus it is possible that many of the unclassified DNA samples that we receive requesting specific genetic tests are inappropriate, which may explain the lower molecular diagnosis rate in this cohort.
Although our clinic receives referrals from neurologists throughout the UK, we also receive referrals directly from general practitioners. The fact that approximately 40% of all CMT patients seen in our clinic have CMT1A indicates that our data are not significantly biased by referral pattern, as this is comparable with population based studies in the UK3 and other clinic based studies.4
Currently, only a limited number of genes can be routinely tested in the UK (http://www.ukgtn.nhs.uk); however, PMP22, GJB1, MPZ and MFN2 testing is widely available. Given that mutations or rearrangements in these four genes account for the vast majority of molecular diagnoses in patients with CMT, with other known genes causing CMT much less commonly, we have suggested an algorithm for genetic testing in the UK based on these results (figure 1). We have kept this algorithm simple by only including the four genes which account for >90% of genetically confirmed CMT. More detailed algorithms are available which include some of the rarer genes (eg, see Saporta and colleagues4); however, since mutations in these rarer genes account for a small proportion of CMT and since there are so many different rare genes known, some having specific phenotypic clues, it may be simpler and more cost effective to refer patients negative for mutations in these four genes for an expert opinion. It is important to emphasise that many patients present without a family history, and this does not necessarily exclude autosomal dominant inheritance. As a molecular diagnosis is significantly more likely to be achieved in patients attending a specialist inherited neuropathy clinic, we suggest that if screening of these four genes does not reveal a causative mutation, the patient should be considered for referral to an inherited neuropathy clinic where a detailed phenotype may give additional clues to guide further genetic testing.
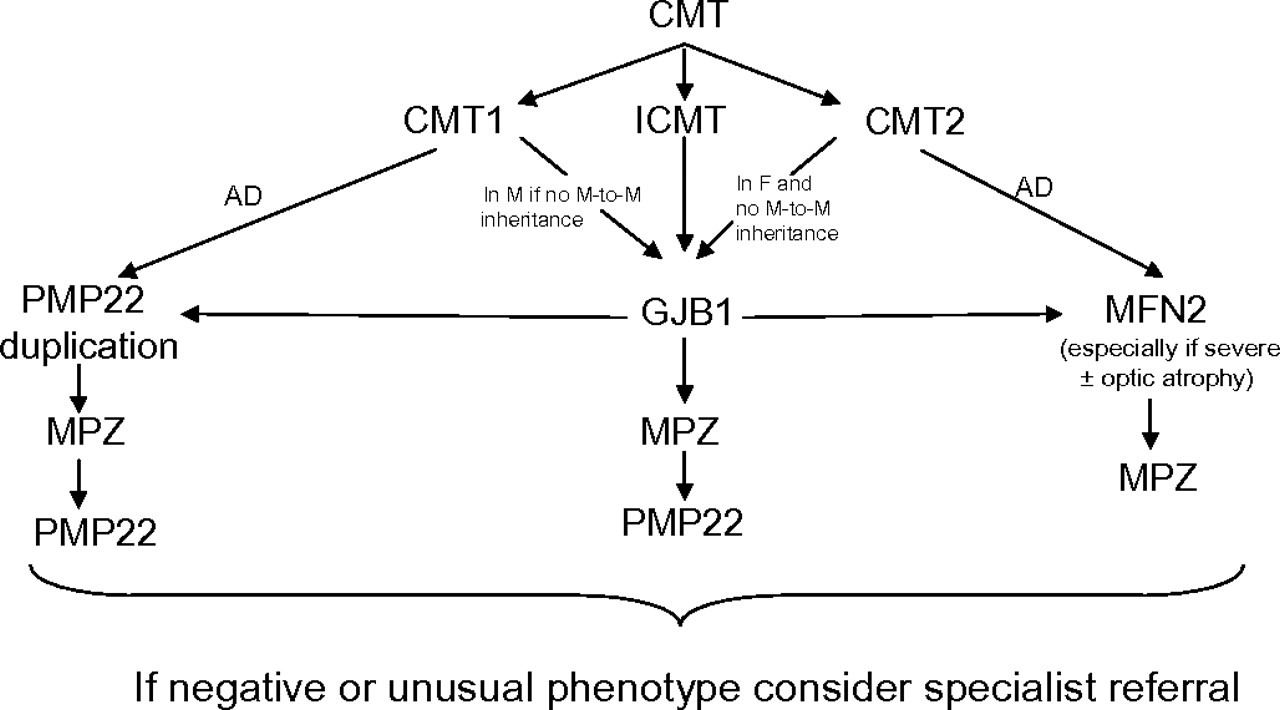
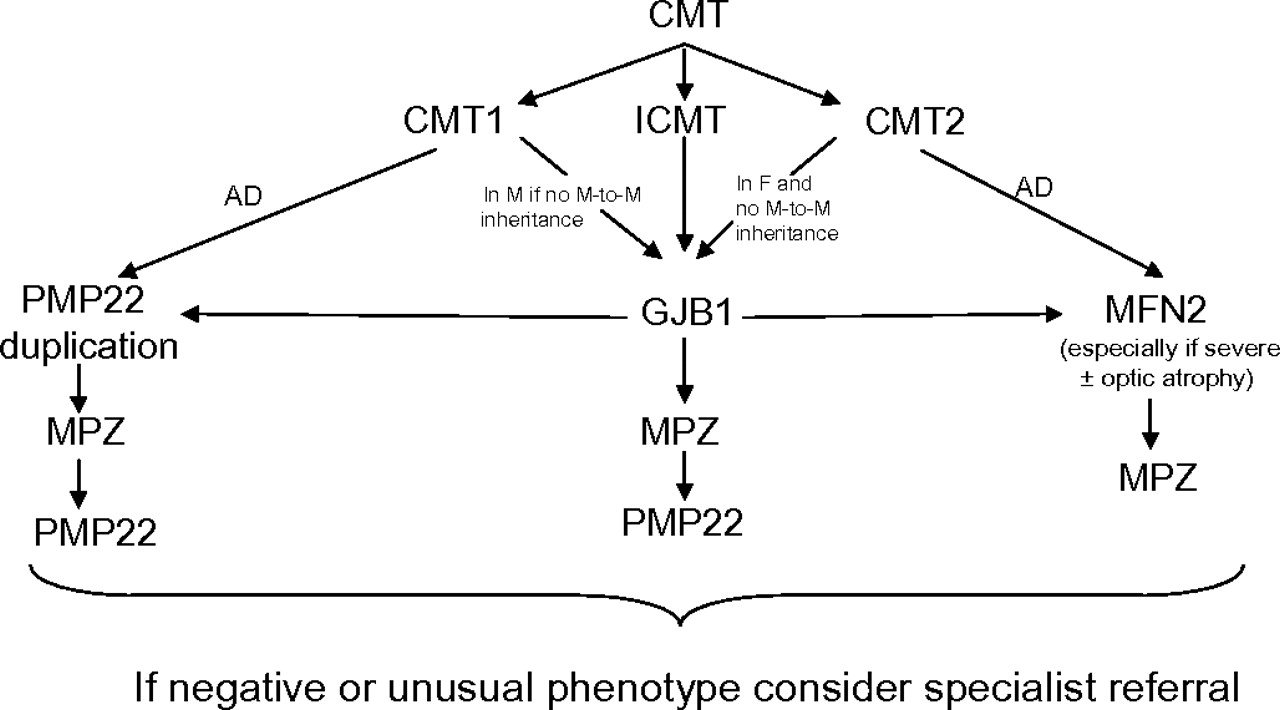{kind=link}
Suggested algorithm for genetic testing of patients with Charcot–Marie–Tooth disease. AD, autosomal dominant; CMT, Charcot–Marie–Tooth disease; CMT1, demyelinating CMT; CMT2, axonal CMT; GJB1, gap junction β-1; ICMT, intermediate CMT; F, female; M, male; MFN2, mitofusin 2; MPZ, myelin protein zero; PMP22, peripheral myelin protein 22.
Conclusion
This study demonstrates that a molecular diagnosis can currently be achieved in over 60% of patients with CMT. A molecular diagnosis is much more likely in patients with CMT1 rather than CMT2, confirming that many genes for CMT2 remain unknown. Four commonly available genes account for over 90% of all CMT molecular diagnoses and we have proposed a diagnostic algorithm based on these results for use in clinical practice. Any patient with CMT without mutations in these four genes or with an unusual phenotype should be considered for referral for an expert opinion to maximise the chance of reaching a molecular diagnosis.
Acknowledgments
The authors would like to acknowledge all the physicians who have sent DNA samples for genetic testing or referred patients to their clinic.
References
Footnotes
Funding MMR is grateful to the Medical Research Council (MRC) and the Muscular Dystrophy Campaign, and SMM, ML and MMR are grateful to the NINDS/ORD (1U54NS065712-01) for their support. Y-TL is grateful to the Ministry of Education, Taiwan, Republic of China, for scholarship support. AP is grateful to CMTUK for funding support. This work was undertaken at University College London Hospitals/University College London, which received a proportion of funding from the Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme.
Competing interests None.
Ethics approval Ethics approval was provided by the National Hospital for Neurology and Neurosurgery Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.