Article Text

Abstract
Objective Inherited ataxias are heterogeneous disorders affecting both children and adults. The primary cause can be identified in about half of the patients and only very few can receive causative therapy.
Methods The authors performed sequencing of known Coenzyme Q10 (CoQ10) deficiency genes in 22 patients with unexplained recessive or sporadic ataxia.
Results CABC1/ADCK3 mutations were detected in four patients and two siblings presenting with cerebellar ataxia, epilepsy and muscle symptoms. Spasticity, dystonia, tremor and migraine were variably present; cognitive impairment was severe in early childhood cases, but was absent in adults. In contrast to previous reports, two of the patients had a later-onset, very mild phenotype and remained ambulatory in their late forties. Muscle biopsy revealed lipid accumulation, mitochondrial proliferation and cytochrome c oxidase-deficient fibres, but no typical ragged red fibres. Respiratory-chain enzyme activities and CoQ10 were decreased in severely affected patients but remained normal in a mildly affected patient at 46 years of age.
Conclusions These observations highlight the importance of screening for a potentially treatable cause, CABC1/ADCK3 mutations, not only in severe childhood-onset ataxia, but also in patients with mild cerebellar ataxia in adult life.
- Autosomal recessive ataxia
- mitochondrial
- coenzyme Q10 (CoQ10) deficiency
- CABC1/ADCK3
- neurogenetics
- mitochondrial disorders
- muscle
- paediatric neurology
- neuromuscular
- HMSN (Charcot–Marie–Tooth)
- muscle disease
- dystrophin
- incl body myositis
- myotonic dystrophy
- muscular dystrophy
- neuropathology
- myasthenia
- myopathy
- biochemistry
- molecular biology
- non-clinician
- metabolic disease
- mitochondrial disorders
- muscle disease
Statistics from Altmetric.com
- Autosomal recessive ataxia
- mitochondrial
- coenzyme Q10 (CoQ10) deficiency
- CABC1/ADCK3
- neurogenetics
- mitochondrial disorders
- muscle
- paediatric neurology
- neuromuscular
- HMSN (Charcot–Marie–Tooth)
- muscle disease
- dystrophin
- incl body myositis
- myotonic dystrophy
- muscular dystrophy
- neuropathology
- myasthenia
- myopathy
- biochemistry
- molecular biology
- non-clinician
- metabolic disease
- mitochondrial disorders
- muscle disease
Introduction
Despite major advances in understanding the genetic forms of ataxia, approximately half of the patients with recessive ataxia do not receive a molecular diagnosis especially in adult life.1 Given the many aetiologies the diagnosis requires the performance of a wide range of analyses, and there is no consensus on the diagnostic value of these examinations.1 Clinical description and evaluation of subtle additional features may provide a key to the precise diagnosis. Apart from vitamin E in the tocopherol-deficient form of ataxia, there are no treatments available to date.
Coenzyme Q10 (CoQ10) deficiencies are a heterogeneous group of autosomal recessive conditions caused by mutations in genes encoding different CoQ10 biosynthesis enzymes.2–4 Ataxia can be the major symptom in patients with CoQ10 deficiency but may also be part of a complex neurological or systemic phenotype.5–8 Autosomal recessive mutations in the CABC1/ADCK3 gene have been identified in nine families to date (16 patients) (table 1).9–11 The frequent association of ataxia and CoQ10 deficiency prompted us to investigate a potentially treatable cause, CoQ10 deficiency, in patients with unexplained recessive or sporadic cerebellar syndrome.
Summary of the previously reported families and our patients with CABC1/ADCK3 mutations
Patients and methods
We studied 22 patients from different families with unexplained ataxia (supplementary material) without a dominant family history, in whom well-known genetic causes of spinocerebellar ataxias (SCA1, 2, 3, 6, 7, 17, DRPLA and Friedreich's ataxia) and other possible causes of a progressive ataxia were excluded by clinical and laboratory investigations (brain MRI, genetic testing, CSF, normal routine biochemistry, blood cell counts, metabolic screening for acyl-carnitines, urinary organic acids, very-long-chain fatty acids, phytanic acid, vitamins E, A, B12, AFP, serum electrophoresis, lipoproteins, lysosomal enzymes, copper, ceruloplasmin, ferritin and iron).
Fifteen patients underwent muscle biopsy. Genetic analyses of mitochondrial DNA (deletions/depletion, some point mutations in muscle DNA) and POLG were normal. Genetic analysis of PDSS1, PDSS2, COQ2, COQ9, CABC1/ADCK3 and APTX was performed in blood DNA in all patients with intronic primers (available for request). In patient 3, a genome-wide SNP-array analysis (Affymetrix Genome Wide Human SNP array 6.0) was performed.
Case reports
Patient 1, an English woman in her forties, was the third child of healthy non-consanguineous parents and had a similarly affected brother. After normal early development, she became clumsy, and her handwriting deteriorated from 15 years of age, followed by a mild gait disturbance, slurred speech, jerky hand movements and swallowing difficulties 3 years later. She had a history of migraine. At age 46, she had a jerky, irregular tremor particularly involving her head and arms, slow ocular pursuits, a subtle dysarthria, mild swallowing difficulties and some muscle fatiguability. Deep tendon reflexes were normal, and she had mildly dystonic posturing and dysdiadochokinesis. Except for a mild bradyphrenia, cognition was normal. At age 46, she remains ambulatory and manages the household. A brain MRI revealed cerebellar atrophy. Her electrophysiology was normal.
Patient 2, a German woman in her fifties, was the first child of healthy non-consanguineous parents. After normal development she had grand mal seizures in childhood and occipital seizures with myoclonic jerks at age 27 years, each preceded by visual auras and migraine. At age 37, she underwent bilateral cataract surgery. At age 46, she had mild cerebellar dysarthria, upper-limb myoclous, dysmetria and gait instability. Eye movements were normal, there was no paresis, but her tendon reflexes were brisk. Cognitive function was normal. She remains ambulatory in her fifties and works full time. A brain MRI revealed prominent cerebellar atrophy (figure 1B). Her electrophysiology was normal.
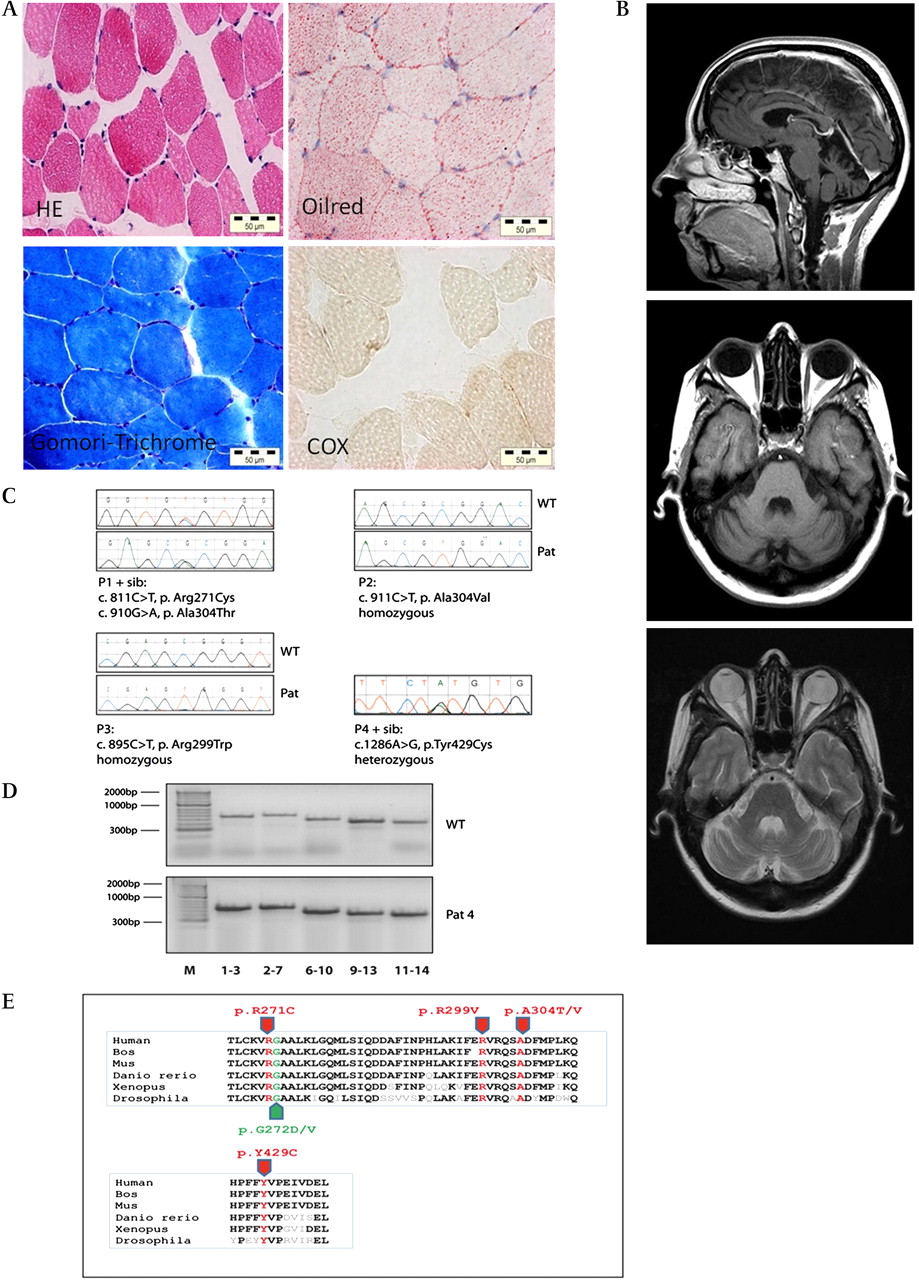
{kind=link}
(A) Muscle histology of patient 2 showing lipid accumulation, subsarcolemmal mitochondrial proliferation and COX-deficient fibres, but no RRF. (B) Brain MRI of patient 2 at age 47 years showed prominent cerebellar atrophy (T1 and T2 sequences). (C) Sequencing results of the detected CABC1/ADCK3 mutations. (D) Analysis of cDNA performed in five overlapping PCR products (exons 1–3, 2–7, 6–10, 9–13 and 11–14) showing normal results in patient 4. All mutations affected highly conserved amino acids. (E) Mutations. Those in red were reported in this paper; those in green were previously described.
Patient 3 was a Norwegian woman in her late teens, the first child of healthy unrelated parents from the same geographical region. Born at term after a normal pregnancy, she sat without support at age 8 months and could walk without support at age 16 months, but her walking remained unsteady. She developed generalised tonic–clonic and absence seizures at 3 years of age, her gait became increasingly atactic, and she gradually lost her ability to walk by 12 years. She learnt to speak but gradually lost her speech from age 14. Owing to severe feeding difficulties, a PEG (percutaneous endoscopic gastrostomy) was inserted at age 13. Presently she sleeps a lot and, when awake, is rather drowsy and has severe mental retardation. Repeated brain MRI investigations at ages 3–16 years showed progressive cerebellar atrophy, enlarged ventricles and thinning of corpus callosum. Her EEG showed epileptiform activity, and her nerve-conduction velocities were normal.
Patient 4 was a woman in her twenties, the first of three children of healthy non-consanguinous parents from Kosovo. One brother was similarly affected. She had normal early development until 18 months of age, when her motor development had become slow. She crawled at 24 months and walked at 3 years of age. During childhood, she developed progressive ataxia, muscle weakness and cognitive impairment, and became wheelchair-bound in her late teens. At age 25, she had horizontal nystagmus, mildly slurred speech, spastic tetraparesis with increased reflexes, severe ataxia, bilateral dysmetria, tremor and depression (wheelchair-bound). Brain MRI showed prominent cerebellar atrophy. Her electrophysiology was normal.
Supplementation with CoQ10 had no beneficial effect on any of the symptoms in patient 1 and 2 after 6 months on 300 mg/day CoQ10. Patients 3 and 4 were recently put on CoQ10 (200 mg/day) without any significant improvement within 2 months.
Results
Morphology and biochemistry of skeletal muscle
Muscle histology revealed mitochondrial proliferation: hyper-reactive fibres on succinate dehydrogenase (SDH) staining, cytochrome c oxidase (COX)-deficient fibres, but no definite ragged-red fibres (RRF) in patients 1, 2 (figure 1A) and 4, with increased lipid accumulation in patients 2 and 4. Respiratory-chain enzyme activities showed combined respiratory chain deficiency, and CoQ10 was significantly decreased in patients 2 and 4 (table 1, supplementary table 3) but was normal in patient 1. We detected decreased CoQ10 in four patients without mutations in the analysed genes.
Molecular genetic studies
Homozygous or compound heterozygous mutations were detected in the CABC1/ADCK3 gene in patients 1–3 (table 1, figure 1C), which showed cosegregation with the phenotype within the families. In patient 3, a genome-wide SNP-array detected a 14.4 Mb region of homozygosity (1q41q42.2 (215 573 596–229 986 845) including CABC1/ADCK3. One heterozygous CABC1/ADCK3 mutation was detected in patient 4, in his similarly affected sibling and the healthy mother, suggesting the possibility of a second mutation from the father. Analysis of cDNA revealed no second abnormality (figure 1D), thus leaving the pathogenicity of this mutation unclear. All detected mutations affect conserved amino acids, predicted to be damaging by PolyPhen (http://genetics.bwh.harvard.edu/pph/) and were absent in 100 control chromosomes.
Discussion
Five major phenotypes of CoQ10 deficiency have been described (encephalomyopathy, multisystem infantile variant, cerebellar form, Leigh syndrome, isolated myopathy).2–4 12 The clinical heterogeneity may reflect blocks at different levels in the complex CoQ10 pathway involving many genes (PDSS1, PDSS2, COQ2, COQ6, COQ9, CABC1/ADCK3).2–4 The number of reported patients carrying mutations in these genes is still low, and no true genotype–phenotype correlations are known, which makes the genetic diagnosis difficult.2–4
After the original descriptions of mutations in CABC1/ADCK3,9 10 only a single study reported two families, and the overall frequency of this disorder remained obscure.11 All reported patients had severe childhood-onset ataxia (<11 years, usually 1–5 years of age) with variable neurological signs and exercise intolerance. The detection rate of CoQ10 deficiency was higher in muscle, than in fibroblasts. The clinical presentation of our patients was similar to the previously reported cases, but two of our patients had adult-onset disease and remained ambulatory in their late forties. Spasticity, dystonia, tremor and migraine were variably present in our patients, and cognitive impairment was severe in early-childhood cases but was absent in adults. None of the cases carrying mutations in CABC1/ADCK3 showed peripheral neuropathy. The symptoms were slowly progressive in most patients, although acute worsening triggered by an episode of epileptic encephalopathy occurred, suggesting mitochondrial aetiology. No patient has died in our cohort, despite having symptoms for up to 31 years, similar to previous reports.9–11
A muscle biopsy revealed lipid accumulation, mitochondrial proliferation and COX-deficient fibres, but typical RRF were absent. Deficiency in CoQ-related assays (complex I+III or II+III) was evident in severely affected patients.13 CoQ10 levels were decreased in muscle, except for patient 1 who had normal levels.
CABC1/ADCK3 encodes a putative kinase, which appears to modulate the biosynthesis of CoQ10. Studies of the yeast homologue, Coq8p, indicate that the protein is required to maintain the stability of Coq3p.14 We detected five novel CABC1/ADCK3 mutations in four families affecting evolutionary conserved amino acids, and the bioinformatic analysis by PolyPhen (http://coot.embl.de/PolyPhen/) indicated, that the mutations are probably damaging (figure 1E). All mutations were absent in 100 normal control chromosomes. Three patients carried compound heterozygous or homozygous mutations. Two different mutations affected the same amino acid (p.Ala304Thr and p.Ala304Val), while p.Arg271Cys was located adjacent to other, previously described pathogenic mutations (p.Gly272Val and p.Gly272Asp). Another mutation (p.Arg299Trp) was detected in a remotely consanguineous Norwegian family, where CABC1/ADCK3 was tested as a possible candidate gene within the single large region of homozygosity. We detected only one heterozygous missense mutation (p.Tyr429Cys) in two siblings, and cDNA analysis revealed no abnormality (figure 1D). Whether we missed a second mutation or whether the disease is related to another cause remains unclear.
The inconsistent effect of CoQ10 supplementation in CABC1/ADCK3 deficiency is not understood. Some reported patients showed a beneficial effect at least temporarily, whereas others did not respond.9–11 Idebenone supplementation resulted in significant worsening in one patient.15 Different CoQ10 derivates may explain the variable effect, but it remains to be determined whether the lack of clinical benefit is related to insufficient doses, incomplete tissue delivery or functional inability.13
Given that, four out of 22 ataxia patients carried mutations in the CABC1/ADCK3 gene, suggesting that this gene defect is a relatively frequent cause of unexplained ataxia after excluding all other aetiologies and should be considered in adult-onset ataxia. Functional studies or animal experiments may be helpful to further explore the disease pathomechanism and a possible beneficial effect of CoQ10 derivates.
Acknowledgments
We thank the patients and their family for contributing to this study.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Download Supplementary Data (PDF) - Manuscript file of format pdf
Footnotes
↵* RH and BC contributed equally to the study.
Funding This study receives funding from the Parkinson's Disease Society (UK), the Medical Research Council (MRC) Translational Muscle Centre, and the UK NIHR Biomedical Research Centre in Ageing and Age-related disease. RH is supported by the Academy of Medical Sciences (UK, BH090164) and by the MRC (UK, G1000848) as part of the MRC Centre for Neuromuscular Diseases.
Competing interests None.
Patient consent Obtained.
Ethics approval Ethics approval was provided by County Durham & Tees Valley 1 Research Ethics Committee (REC 08/H0905/106).
Provenance and peer review Not commissioned; externally peer reviewed.