Article Text

Abstract
Sporadic cerebral amyloid angiopathy (CAA) is a common age related cerebral small vessel disease, characterised by progressive deposition of amyloid-β (Aβ) in the wall of small to medium sized arteries, arterioles and capillaries of the cerebral cortex and overlying leptomeninges. Previously considered to be a rare neurological curiosity, CAA is now recognised as an important cause of spontaneous intracerebral haemorrhage and cognitive impairment in the elderly, two fundamental challenges in the field of cerebrovascular disease. Our understanding of the pathophysiology and clinical manifestations of CAA continues to evolve rapidly, with the use of transgenic mouse models and advanced structural and/or molecular neuroimaging techniques. Yet, despite remarkable recent interest, CAA remains under-recognised by neurologists and stroke physicians. In this review, a fresh look at key developments in understanding the complex pathophysiology, important clinical and radiological features, diagnostic approaches and prospects for rational therapies for this enigmatic small vessel disorder is provided.
Statistics from Altmetric.com
Introduction
Sporadic cerebral amyloid angiopathy (CAA) is a common small vessel disease of the brain, characterised by the progressive deposition of amyloid-β (Aβ) protein in the walls of small to medium sized arteries (up to about 2 mm in diameter1), arterioles and capillaries in the cerebral cortex and overlying leptomeninges.2 3 CAA can also affect cerebellar vessels but only rarely those in the brainstem or basal ganglia. Although known to pathologists for over a century,4 5 CAA was not linked to clinical disease until as late as the 1960s when it was suggested to be a rare cause of intracerebral haemorrhage (ICH).6–8 In recent years, CAA has been ‘rediscovered’ as a common and important cause of spontaneous ICH, which remains the most devastating form of stroke, with a death rate approaching 50% in contrast with improved outcomes from ischaemic stroke.9 10 An increased understanding of CAA thus holds promise for improved prevention and treatment of ICH.
The growing interest in CAA is at least partly thanks to two fields of research, which have been important in defining the expanding clinical–radiological phenotype and the underlying pathophysiology of the disease: (1) neuroimaging, which now allows an unprecedented ability to investigate CAA dynamics in vivo using MRI to reveal complex patterns of cerebral bleeding (including lobar microbleeds11) and ischaemia, and an increasing repertoire of specific amyloid binding ligands3 12–16; and (2) transgenic mouse studies, which have allowed the experimental alteration of amyloid peptide expression and molecular structure, providing significant mechanistic insights. Despite these advances, CAA remains under-recognised by neurologists and stroke physicians, making a fresh look especially timely. In this review (see box 1 for search strategy), we provide a comprehensive update, emphasising the widening spectrum of CAA clinical presentations and neuroimaging features, including diagnostic approaches to reliably identify the disease in vivo. Finally, we discuss improved prospects for rational preventive or disease modifying therapies for this common and devastating microangiopathic disorder.
Search strategy and selection criteria
References were identified through PubMed with the search terms: ‘cerebral amyloid angiopathy’; ‘microbleed(s) or microh(a)emorrhage(s) and cerebral amyloid angiopathy’; ‘intracerebral h(a)emorrhage’; and ‘vascular cognitive impairment’ between January 1970 and August 2011. The references from identified articles and the authors' own files were also searched for relevant publications. Only papers published in English were reviewed. The final reference list was chosen on the basis of relevance to the topics covered in this article.
Epidemiology and risk factors
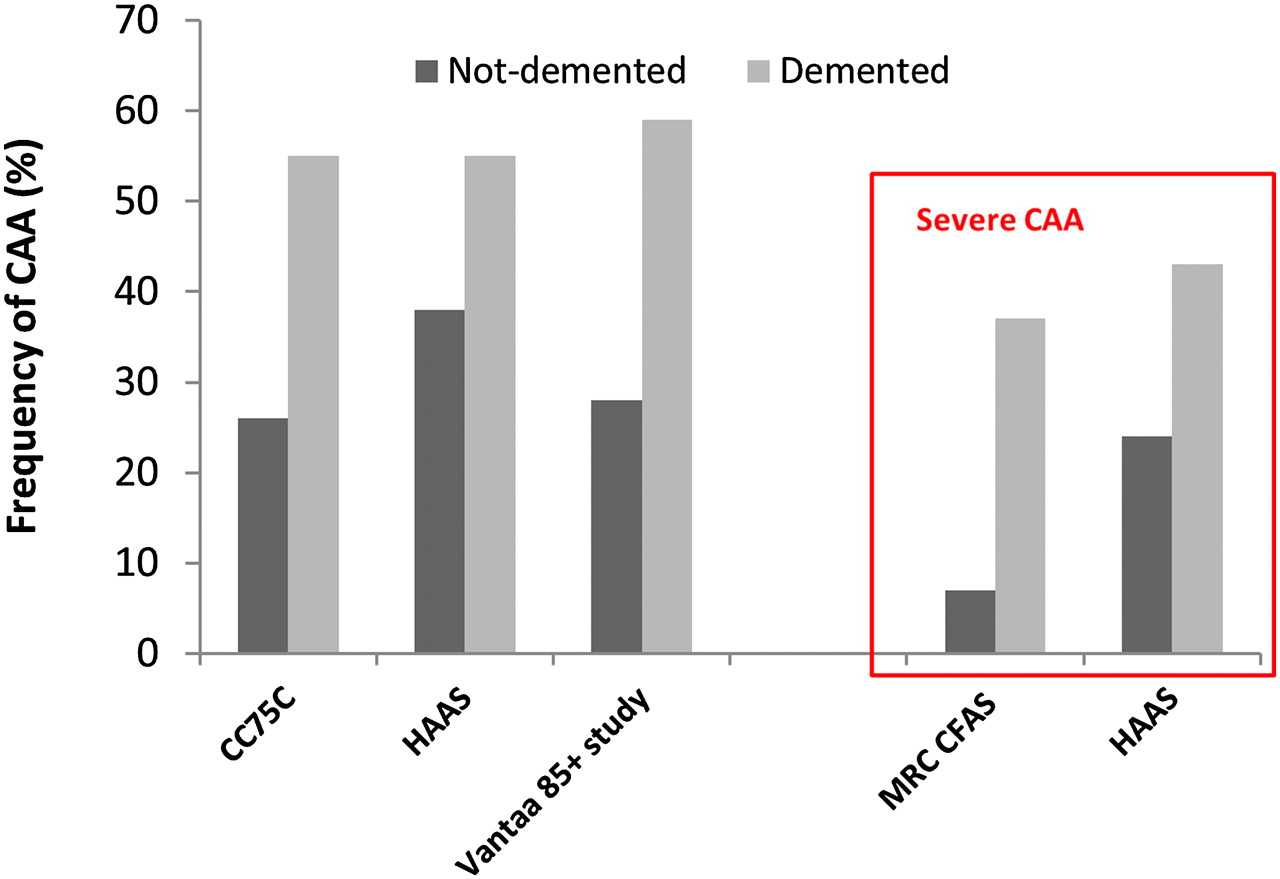
Pathologically defined CAA is common in the elderly.17–20 Population based autopsy studies indicate a CAA prevalence of 20–40% in non-demented and 50–60% in demented elderly populations (figure 1).19 21–24 Furthermore, CAA pathology may be severe in older individuals (figure 1): in the Honolulu–Asia Ageing Autopsy Study, severe CAA was found in 43% of demented and 24% of non-demented elderly individuals (mean age at death 85 years).23 In Alzheimer's disease (AD), CAA is almost invariable being found at autopsy in more than 90% of cases.17 25 However, most of these patients have mild CAA; severe CAA is found in about 25% of AD brains.26

The frequency of cerebral amyloid angiopathy (CAA) in demented and non-demented elderly individuals in population based clinicopathological studies. Note the increased prevalence of CAA, even if only severe pathology is taken into account. CC75C, Cambridge City over 75 Cohort21; HAAS, Honolulu–Asia Ageing Study23; Vantaa 85+ study24; MRC–CFAS, MRC Cognitive Function and Ageing Study.22
Advancing age is the strongest known clinical risk factor for developing CAA.2 In a community based sample of 100 individuals, the prevalence of cortical vascular Aβ deposition progressively increased from the seventh to the ninth decades,27 a pattern also observed in 784 consecutive autopsies, corrected for over-representation of AD.28 Moreover, patients with CAA related ICH (suggesting advanced disease) in large autopsy series were all older than 60 years (and most over 70 years of age).7 29 30 Sporadic CAA is seldom reported before the sixth decade of life; occasional patients presenting in their 50s have been described.31
In contrast with hypertensive arteriopathy—the other main form of small vessel disease and cause of ICH32—the risk of CAA is not accounted for by conventional cardiovascular risk factors other than age.2 Hypertension is not considered a risk factor for developing CAA but may increase the risk of CAA related ICH. Vinters2—in a clinicopathological series of 107 pathologically proven CAA cases—found the prevalence of hypertension to be around 32%, similar to community dwelling elderly populations,33 while another pathological study reported that CAA patients with ICH were more frequently hypertensive (50%) than those without ICH (23%), suggesting that hypertension may contribute to CAA related cerebral bleeding.34 In a recent multicentre cohort of patients with spontaneous ICH, we found that the prevalence of hypertension in CAA related ICH was 62%—significantly less than in non-CAA related ICH (85%).35 Whether hypertension in association with CAA confers a greater risk for ICH compared with CAA alone is an important clinical question.36–38 Evidence from the PROGRESS trial of blood pressure lowering after stroke showed that a mean blood pressure reduction of 9/4 mm Hg reduced the risk of future CAA related ICH by about 77%, supporting an important causal role for hypertension.38
Apolipoprotein E (ApoE) alleles are the only known genetic risk factors for sporadic CAA.39 ApoE is a protein with crucial roles in lipoprotein complexes, which regulate lipid metabolism by binding to cell surface receptors and proteins associated with lipid transfer and lipolysis.39 There are three major polymorphisms in the ApoE gene—namely, ɛ4, ɛ2 and ɛ3—resulting in a single amino acid change40 which dramatically alters the functional properties of ApoE isoforms.41 These alleles have a strong dose dependent effect on the risk of developing CAA and its clinical severity. Thus ApoE ɛ4 in both postmortem and clinical series increases the risk of sporadic CAA related lobar ICH; moreover, the number of ɛ4 alleles relates to clinical severity.39 42–44 Individuals carrying the ApoE ɛ2 allele also have an increased risk of CAA related lobar ICH.44 45 Both of these risk alleles are also associated with a younger age of first ICH,46 greater likelihood of haematoma expansion, poorer clinical outcome47 48 and a higher risk of recurrence.49 Furthermore, the two allelic variants interact: patients with both ApoE ɛ2 and ɛ4 alleles have the earliest disease onset and highest risk of early ICH recurrence.49 50 The ɛ2 and ɛ4 alleles might promote CAA related haemorrhage through distinct mechanisms: ɛ4 by promoting Aβ deposition and ɛ2 by inducing structural changes in amyloid laden vessels, making them prone to rupture.47 48 50–52 Other as yet unidentified genetic polymorphisms relating to amyloid metabolic pathways (figure 2A) may also play a role in sporadic CAA, (eg, presenilin-1, neprilysin and transforming growth factor β-1),57–59 and are a topic of ongoing investigation.

(A) Amyloid-β (Aβ) production, elimination and deposition in cerebral amyloid angiopathy (CAA). Converging evidence indicates that the major source of Aβ is neuronal. It is generated by sequential cleavage of amyloid precursor protein (APP) by β- and γ-secretases, in proportion to neuronal activity. Aβ is eliminated from the brain by four major pathways: (a) proteolytic degradation by endopeptidases (such as neprilysin and insulin degrading enzyme (IDE)); (b) receptor mediated clearance by cells in the brain parenchyma (microglia, astrocytes and to a lesser extent neurones); (c) active transport into the blood through the blood–brain barrier (BBB); (d) elimination along the perivascular pathways by which interstitial fluid drains from the brain.53 54 Specialised carriers (eg, ApoE) and/or receptor transport mechanisms (eg, the low density lipoprotein receptor (LDLR) and LDLR related protein (LRP1)) are involved in all major cellular clearance pathways. Vascular deposition is facilitated by factors that increase the Aβ40:Aβ42 ratio (while increased Aβ42 leads to oligomerisation and amyloid plaques) and impede perivascular passage. As the clearance mechanisms fail with age, Aβ is increasingly entrapped from the perivascular drainage pathways into the basement membranes of capillaries and arterioles of the brain leading to CAA.55 56 ApoE alleles have a differential effect on different molecular and cellular processes of Aβ production, elimination and deposition in a way that they either increase or decrease the risk of developing CAA. (B) The roles of different ApoE alleles in various pathways in the brain which might contribute in the pathogenesis and pathogenicity of CAA.
Neuropathology
Morphological characteristics, natural history and severity grading
CAA primarily involves neocortical and leptomeningeal arterioles, to a lesser extent capillaries and, very rarely, venules.3 In contrast with amyloid plaques found in AD—which are predominantly composed of the 42 amino acid residue fragment (Aβ42)—the vascular amyloid in CAA is mostly composed of the more soluble, 40 amino acid fragment (Aβ40), suggesting different pathophysiological mechanisms for pathological deposition (see below).60–63 Cerebral vessels with moderate to severe CAA show an acellular wall thickening with a strongly eosinophilic smudgy appearance on haematoxylin–eosin stained sections.64 Congo red staining, under polarised light, reveals amyloid deposits as ‘apple green’ birefringence (hence the term congophilic angiopathy)2 65 although immunological stains for Aβ are highly specific and now widely used (figure 3). The development of CAA is progressive, with Aβ first appearing in the abluminal aspect of the tunica media, surrounding smooth muscle cells, and in the adventitia (figure 3).2 At the initial stage, the vessel wall structure is intact, but as the disease progresses, there is pan-mural amyloid accumulation and loss of smooth muscle cells.3 In severe CAA, detachment and delamination of the outer part of the tunica media result in the so-called ‘double barrel’ appearance (figure 3)3; fibrinoid necrosis and microaneurysm formation also occur in advanced disease. There may also be microbleeding with perivascular deposition of erythrocytes and blood breakdown products.64 Endothelial cells are usually preserved even in vessels severely affected by CAA.66 Occasionally Aβ is deposited in the surrounding brain parenchyma immediately adjacent to an affected vessel (sometimes called ‘dyshoric CAA’).

Histopathological features of cerebral amyloid angiopathy (CAA). (A1–A3) Morphological changes of the vessel walls of leptomeningeal arterioles, as revealed by haematoxylin–eosin staining. In mild and moderate CAA, only minimal structural changes can be detected: in (A2) the arrowhead points to amyloid deposition in the vessel wall. However, in advanced CAA, there are significant structural alterations, the most extreme of which is double barrelling (detachment and delamination of the outer part of the tunica media; bracket in (A3)). (B1–B3) A similar pathological range of CAA related changes in leptomeningeal arterioles using immunohistochemical detection of Aβ. In mild CAA (B1), there is patchy deposition of amyloid in the vessels wall. Moderate CAA shows more dense amyloid deposition which spans the entire vessel wall (B2) while severe CAA shows double balled vessels and endothelial involvement (B3). (C1–C3) Pathological findings of CAA in cortical arterioles. C2 shows moderate CAA with pan-mural deposition of Aβ along with Aβ deposition in the surrounding brain parenchyma (arrowhead). In (C3), a double barrel vessel can be seen although this is less common compared with leptomeningeal vessels.
CAA is also associated with cerebral ischaemic damage,17 26 67 68 including cortical microinfarcts,69 and white matter pathology (demyelination and gliosis).8 17 62 Microinfarcts are predominantly lobar (cortical–subcortical), usually in patients with severe CAA. One possible mechanism for these ischaemic lesions is occlusion or reduced perfusion in amyloid laden cortical vessels affected by CAA.
The changes described above provide the basis of neuropathological scoring systems for CAA,34 67 70 each with strengths and limitations.71 No standardised consensus neuropathological criteria for rating CAA are available72 but are desirable to allow comparison of CAA pathological studies between centres. A more detailed discussion of CAA severity grading can be found in a recent review by Attems and colleagues.3
Pathological subtypes of sporadic cerebral amyloid angiopathy
At least two distinct pathological subtypes of CAA have been described: CAA type 1, characterised by Aβ in cortical capillaries (with or without involvement of other vessels)3; and CAA type 2, where Aβ deposits are restricted to leptomeningeal and cortical arteries, arterioles and, rarely, veins.73 Aβ deposition in the wall of capillaries (capillary CAA) may cause luminal obstruction in the most severe stages.1 The Apo E ɛ4 allele is most strongly associated with CAA type 1 while Apo E ɛ2 is more associated with CAA type 2.73 CAA type 1 appears to be more closely associated with parenchymal amyloid deposition in AD.74
Topographical distribution
Sporadic CAA favours posterior cortical regions; the occipital lobe is most frequently affected, followed by the frontal, temporal and parietal lobes.2 3 The occipital lobe is also most severely affected.75 76 The cerebellum can be affected in advanced stages while the basal ganglia, thalami, white matter and brainstem are typically spared.71 The distribution of CAA pathology shows a characteristic patchy pattern,2 so that foci of vessels severely affected by CAA may be adjacent to other with mild or absent Aβ deposition.2 3 The practical consequence of this is that cerebral biopsy may miss patchy CAA pathology.
Pathophysiological pathways
Amyloid-β production, clearance and accumulation
Aβ is generated by sequential cleavage of amyloid precursor protein (APP) by β- and γ-secretases. Mutations in the gene encoding the APP account for some rare (usually autosomal dominant) forms of CAA, including CAA Dutch type.77 Familial non-Aβ forms of CAA include familial British dementia,78 79 familial Danish dementia80 and Icelandic cystatin C mutation.81 In general, hereditary forms of CAA have an earlier onset and more severe clinical manifestations than sporadic CAA.64 82 Although exceptionally rare, familial CAAs have provided significant insights on how mutations in the coding region of the APP contribute to CAA pathogenesis: for example, the Iowa, Dutch, Italian and Arctic mutations render Aβ highly toxic to vessel wall components83–85 and more resistant to proteolytic degradation86 or clearance from the brain (figure 2).55
Factors that initiate or promote Aβ peptide deposition in the much more common sporadic CAA are not as well understood. Nevertheless, transgenic mouse models of cerebral amyloid deposition3 have provided the following insights (figure 2): (1) the major source of human Aβ is neuronal87 88; (2) an increased ratio of Aβ40:Aβ42 in the brain results in a shift from brain parenchyma to the vasculature (perhaps by increasing the solubility of Aβ and thus its diffusion into the vessel wall)53; and (3) vascular Aβ deposition largely results from impaired clearance of Aβ (rather than overproduction), especially along perivascular drainage pathways.3 54 89 Impairment of perivascular drainage pathways has emerged as a key mechanism in sporadic CAA3 56 90: these efflux channels may be conceptualised as a cerebral ‘lymphatic system’, allowing interstitial fluid and solutes to drain out of the brain along basement membranes in the capillary walls, and between smooth muscle cells in the tunica media of small arteries (in the opposite direction to arterial blood flow) (figure 2).91 This transport system is thought to be driven by pulsations of the blood vessel wall.91 92 As this and other clearance mechanisms fail in the ageing brain, or under other pathological conditions, Aβ is increasingly trapped and deposited in the walls of small arteries (figure 2).91 Evidence is emerging that cerebrovascular disease may impede the drainage along the perivascular pathways, contributing to CAA pathogenesis.90 93
It has been suggested that Aβ deposition could further impair/block the perivascular drainage, leading to dilation of perivascular spaces (also known as Virchow–Robin spaces), not only within lobar regions but also in the underlying white matter that itself is unaffected by CAA.94 95 These enlarged perivascular spaces can reach several millimetres in diameter and may be visible on appropriate brain imaging; this requires further investigation as a potential useful neuroimaging marker of CAA.95 96
As we have seen, ApoE is a strong genetic risk factor for CAA, an effect mediated by its important role in Aβ metabolism, aggregation and clearance (figure 2B).39 54 89 ApoE ɛ4 increases the Aβ40:Aβ42 ratio, shifting amyloid deposition to the vessels instead of brain parenchyma,53 and may reduce the efficiency of efflux of Aβ along perivascular channels,3 97 influencing CAA risk and age of onset.39 98 ApoE genotype may also interact with other small vessel disease changes: hypertensive arteriopathy, which leads to stiffening of the vessel wall, may reduce the pulsatile driving movements required for efficient perivascular drainage and thus contribute to the risk of CAA.3
From amyloid-β deposition to cerebral amyloid angiopathy pathogenesis
Aβ deposition has complex effects on vascular structure and function which can result in brain injury.32 99 Important morphological changes include: loss of smooth muscle cells100; vessel wall thickening and lumen restriction67; endothelial dysfunction; and a loss of compliance leading to brittle, fragile vessels prone to microaneurysm formation and leakage.3 Acute trigger factors—for example, sudden increases in blood pressure—or minor trauma (regularly encountered in clinical practice but not to our knowledge formally studied) may cause the rupture of these abnormally weak, amyloid laden vessels. Aβ deposition may also impair local regulation of cerebral blood flow,99 neurovascular unit function101 and general homeostatic mechanisms in the ageing brain.35 Other effects of vascular Aβ, including blood–brain barrier disruption and active inflammation, could also contribute.3 99 Moreover, even without vascular deposition, soluble Aβ can cause abnormal vascular reactivity99 and induce the activation of inflammatory mediators, including matrix metalloproteinase-9 and -2.3 102 103
The expanding clinical spectrum of sporadic cerebral amyloid angiopathy
There are at least four important clinical presentations associated with CAA:
Symptomatic intracerebral haemorrhage
Cognitive impairment and dementia
Rapidly progressive cognitive and neurological decline
Transient neurological symptoms
Intracerebral haemorrhage
Association between cerebral amyloid angiopathy and intracerebral haemorrhage
CAA is most often recognised in life by symptomatic, spontaneous, lobar ICH in elderly patients. The majority of ICHs (>75%) in the elderly are classified as spontaneous (sometimes also termed primary or non-traumatic), resulting from rupture of small arteries affected by two main processes: hypertensive arteriopathy or CAA. Hypertensive arteriopathy—characterised by lipohyalinosis and fibrinoid necrosis of small lenticulostriate arterial perforators—is considered an important cause of spontaneous ICH in deep or infratentorial locations (basal ganglia, thalamus and pons). By contrast, CAA related ICHs preferentially affect cortical–subcortical (lobar) regions (especially the occipital and temporal lobes104), less commonly the cerebellum and rarely deep or brainstem structures, reflecting the distribution of the underlying microangiopathy.2 30 75 The predilection for the occipital lobes is not well understood but one hypothesis is that greater tortuosity of occipital small arteries impairs perivascular drainage.3
Clinicopathological studies suggest that CAA related ICH accounts for at least 5–20% of all spontaneous ICH,2 17 26 34 105 106 and that the link is strongest for lobar ICH. However, there are methodological challenges in attributing ICH to CAA: most pathological case control studies did not systematically control for potential confounding risk factors for CAA, including cognitive impairment, ethnicity or age. Furthermore, pathological studies showed differences in the prevalence of ICH only when comparing the presence of low grade CAA versus moderate to high grade CAA,23 26 34 67 107–110 suggesting that mild CAA may not confer such a high risk of ICH. Since many elderly individuals in population based studies have subclinical CAA without haemorrhage, CAA (especially if mild) may not be a sufficient cause of lobar ICH alone, but may interact with other factors—for example, hypertension, neurodegenerative pathology or the use of anticoagulant drugs.23 26 34 67 107–110
Clinical features of cerebral amyloid angiopathy related intracerebral haemorrhage
CAA related ICHs have some distinct neuroimaging features, which are shown in figure 4.75 111 However, the clinical presentation of CAA related ICH is similar to other forms of lobar ICH (eg, due to tumours or arteriovenous malformations) and varies according to ICH size and location. Patients usually present with an acute stroke syndrome with focal neurological deficits that may be associated with headache, nausea, vomiting, seizures and/or altered level of consciousness (especially large lobar bleeds).10 There may also be a history of apparently minor head trauma, which might predispose to ICH in individuals with CAA. The typical lobar location of haemorrhage more often leads to acute seizures than in deep ICH. A first ever ICH due to CAA may be relatively mild clinically but this is counterbalanced by the high risk of recurrent haemorrhages; indeed, subsequent ICH (which characteristically may cluster over a short period of time (days to weeks)) is often much more severe.112 In the longer term, survivors of lobar ICH are at higher risk of recurrence compared with deep ICH, with a rate of about 10% per year in elderly cohorts.2 112 Recurrent haemorrhages are typically lobar, often in the same lobe as the initial CAA related bleed.104 Multiple simultaneous lobar haemorrhages are characteristic of CAA related ICH. Recovery from lobar ICH is often poor: negative prognostic factors include older age46 and larger haematoma size113; conversely, a small superficial ICH without intraventricular extension is associated with better outcome.
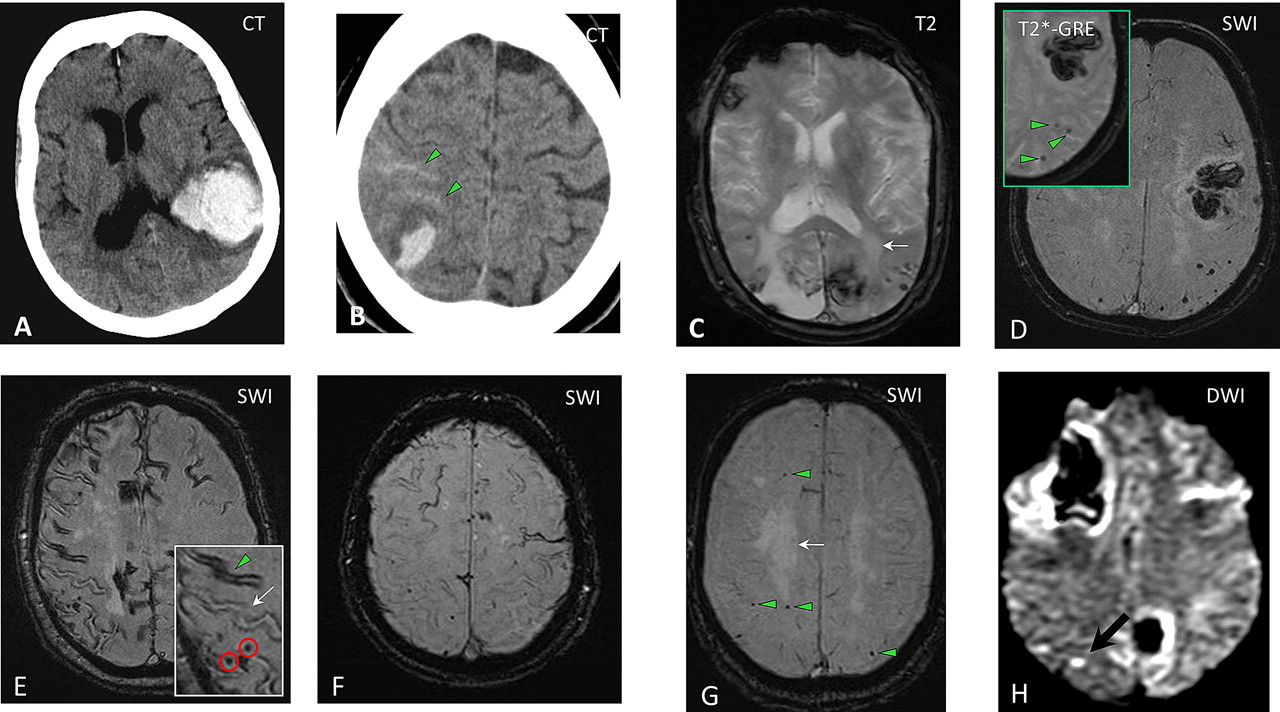
The spectrum of imaging manifestations of sporadic cerebral amyloid angiopathy (CAA). (A) An acute lobar haematoma on CT scan. Some extension of the bleeding in the posterior horn of the left ventricle can be seen. (B) CT scan of a patient with a small posterior cortical haematoma. Acute cortical subarachnoid haemorrhage (cSAH) is evident in two adjacent sulci (arrowheads). (C) A T2 weighted MRI of a patient with probable CAA showing two lobar foci of recent/subacute intracerebral haemorrhage (ICH): in the medial aspect of the left occipital lobe and in the right inferior frontal gyrus. There is also a large old lobar haemorrhage involving the right occipital lobe, some scattered cortical–subcortical cerebral microbleeds (CMBs) in posterior brain regions, as well as confluent white matter hyperintensities in the posterior white matter (leukoaraiosis: arrow). (D) Susceptibility weighted imaging (SWI) and T2* weighted gradient recalled echo (T2*-GRE) (inset) done on the same day in a patient with a lobar haemorrhage. The detection of strictly lobar CMBs (better demonstrated on SWI) is consistent with a diagnosis of probable CAA. (E–F) cSAH (linear hypointensities in the subarachnoid space on T2*-GRE/SWI) and cortical superficial siderosis (hyperintense on T2*-GRE/SWI). The inset in (E) demonstrates the coexistence of cSAH (arrowhead), focal cortical siderosis in an adjacent sulcus (arrow) and some CMBs (circles). Focal cortical siderosis represents the chronic lesion following acute cSAH. (G) SWI in a patient presenting with progressive cognitive impairment led to the detection of multiple strictly lobar microbleeds, characteristic of CAA. Confluent white matter changes (arrow) are also visible. (H) Diffusion weighted imaging (DWI) showing a small acute ‘silent’ ischaemic lesion in the right parietal lobe (arrow) in a patient with probable CAA.
Anticoagulant related haemorrhage
CAA may be an important risk factor or cause for ICH related to oral anticoagulation use. Over the past decade there has been a fivefold increase in the incidence of anticoagulant related ICH, which now accounts for about 15% of all ICH.114 This trend is probably due to increasing use of warfarin to prevent cardioembolic stroke in elderly patients with atrial fibrillation. Anticoagulant use per se should not cause ICH if cerebral vessels are intact but the presence of CAA, rendering vessels brittle and fragile, is a plausible aggravating factor for such haemorrhage; an otherwise innocuous minor and self-limiting vessel leak (eg, a cerebral microbleed (CMB), see below) could form a life threatening haematoma if the leaking vessel is damaged by advanced CAA. Evidence supporting a link between CAA and anticoagulation related ICH includes the following observations: first, most such ICH occur with international normalised ratios within the therapeutic range115 suggesting that an intrinsic disorder of cerebral small vessels could be important; and second, the ApoE e2 allele is more common in warfarin related ICH than in patients on warfarin without ICH, supporting a role for CAA.115 Although CAA may underlie a substantial proportion of anticoagulation related haemorrhages, prospective studies with reliable diagnosis of CAA in life (eg, by MRI evidence of lobar CMBs or molecular imaging) in cohorts of patients treated with anticoagulants are urgently needed to answer this question (one large prospective MRI study is currently underway in the UK: http://www.ucl.ac.uk/cromis-2).
CAA may also be a risk factor for ICH after thrombolysis: spontaneous CAA related and thrombolysis related haemorrhages share some features, including a predilection of lobar brain regions, multiplicity of haemorrhages, age dependency and an association with dementia and leukoaraiosis.116 In one small study, two of five cases of ICH after thrombolysis for acute myocardial infarction had severe CAA identified.117
Cognitive impairment and dementia
There is now increasing evidence that CAA is an important contributor to cognitive impairment72 118 although dissecting its independent cognitive impact is confounded by the presence of coexisting AD and other age related pathologies (eg, hypertensive arteriopathy). Nevertheless, in population based clinical–pathological studies, the prevalence of CAA is consistently higher in demented compared with non-demented patients (figure 1).19 In the population based Medical Research Council–Cognitive Function and Ageing Study, CAA was significantly associated with dementia (OR 9.3, 95% CI 2.7 to 41.0), even after controlling for age and dementia related neuropathologies (eg, neuritic and diffuse plaques).22 Similarly, the Honolulu–Asia Ageing Autopsy Study revealed a significantly higher prevalence of severe CAA in demented versus non-demented patients (43% vs 24%) (figure 1).23 CAA may worsen the severity of cognitive dysfunction in AD: CAA together with AD pathology has been associated with significantly worse cognitive performance during life, compared with AD alone, even after controlling for age, neurofibrillary tangles and amyloid plaques number, infarctions and ApoE genotype.23 There are few studies of the specific pattern of cognitive impairment associated with CAA; a recent autopsy series found that moderate to severe CAA (present in 19% of the study population) was associated with lower performance in specific cognitive domains, notably perceptual speed and episodic memory, after accounting for AD pathology and other potential covariates.119 The pathophysiological mechanisms by which CAA could cause cognitive impairment have not been well established118 but relevant lesions on brain imaging could include cerebral microbleed,120 microinfarcts35 121 and white matter changes.122
CAA is thus emerging as a potentially important link between neurodegenerative and cerebrovascular pathology.123 Vascular cognitive impairment and AD are now conceptualised as a continuum118 124 125 with complex interactions and shared risk factors.99 123 CAA seems likely to exacerbate the deleterious effect of neurodegenerative pathology on the brain, lowering the threshold for overt dementia.99 118 Unravelling the independent contribution of CAA to cognitive function is particularly important as it could lead to new therapeutic strategies.
Rapidly progressive cognitive and neurological decline: cerebral amyloid angiopathy related inflammation
CAA is clearly a direct cause of cognitive impairment in the uncommon but clinically striking presentation of CAA related inflammation (also termed cerebral amyloid angiitis, amyloid β related angiitis and cerebral amyloid inflammatory vasculopathy).126 CAA related inflammation typically affects older adults, who present with acute to subacute cognitive decline, headache, behavioural change, seizures and focal neurological deficits.126 Typical MRI findings include patchy or confluent, asymmetric T2 weighted or FLAIR white matter hyperintensities (with or without mass effect and leptomeningeal or parenchymal enhancement).126 T2* weighted gradient recalled echo (T2*-GRE) or susceptibility weighted imaging (SWI) may reveal previous lobar haemorrhage and/or multiple cortical and subcortical microbleeds.126 The major differential diagnoses include infections (in particular progressive multifocal leucoencephalopathy), neurosarcoidosis, immune related conditions (eg, acute disseminated encephalomyelitis)127 and malignancies.126 Definite diagnosis requires brain and leptomeningeal biopsy showing perivascular inflammation with mononuclear or multinucleated giant cells associated with Aβ laden vessels and/or frank vasculitis.126 Although the clinical course of CAA related inflammation is varied, it is important to recognise because it may respond well to immunosuppressive treatment (eg, high dose corticosteroids or cyclophosphamide).126 128 This distinct syndrome has parallels with that observed in patients with AD who developed meningoencephalitis after immunisation against human Aβ, where postmortem examination revealed inflammation and/or vasculitis associated with CAA.129 130
Transient focal neurological episodes
After ICH, the next most commonly described clinical presentation of sporadic CAA is with transient neurological episodes,131–133 sometimes termed ‘amyloid spells’. The most common type of attack involves recurrent, stereotyped episodes of ‘positive’ spreading sensory symptoms (paraesthesias).131 132 Although there are a number of small case reports and series,131 132 134 135 no large systematic studies have investigated the prevalence or semiology of these phenomena. At least two other types of transient events have been described: partial motor seizure-like episodes (eg, limb shaking); and visual disturbances (usually positive visual symptoms similar to migrainous auras). Spells are typically brief, almost always less than about 30 min, and usually less than a few minutes. The attacks seem likely to be related to haemorrhagic components of CAA: associated neuroimaging findings reported include CMBs and convexity subarachnoid haemorrhage (cSAH) in the cortical region corresponding to the spell (figure 4E).131 135 The diagnosis of these CAA related attacks is of clinical relevance as they seem to precede serious symptomatic ICH in some patients; antiplatelet or anticoagulant use following such an attack misdiagnosed as a transient ischaemic attack (TIA) could therefore cause potentially avoidable intracranial bleeding. The underlying mechanisms of CAA transient spells remain unclear but could include seizure-like activity (perhaps related to small areas of bleeding—for example, microbleeding, cSAH or superficial siderosis); a direct effect of amyloid or bleeding on local cortical function; or spreading cortical depression.131 The responsiveness of these attacks to antiepileptic drugs as well as their spreading nature in many of the reported cases is in favour of a seizure-like mechanism for their pathophysiology. In a case series by Roth and colleagues,132 four out of six patients with these transient attacks responded to anticonvulsants while the other two patients showed improvement after cessation of antiplatelet therapy. Typical TIA-like episodes have also been reported in CAA133 but whether these are genuinely due to ischaemia and should be treated with antithrombotic agents requires further study.
Neuroimaging (MRI) correlates of cerebral amyloid angiopathy
The important MRI correlates of CAA (figures 4 and 5) include:
Cerebral microbleeds
White matter changes (leukoaraiosis)
Convexity subarachnoid haemorrhage
Cortical superficial siderosis
Silent acute ischaemic lesions
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
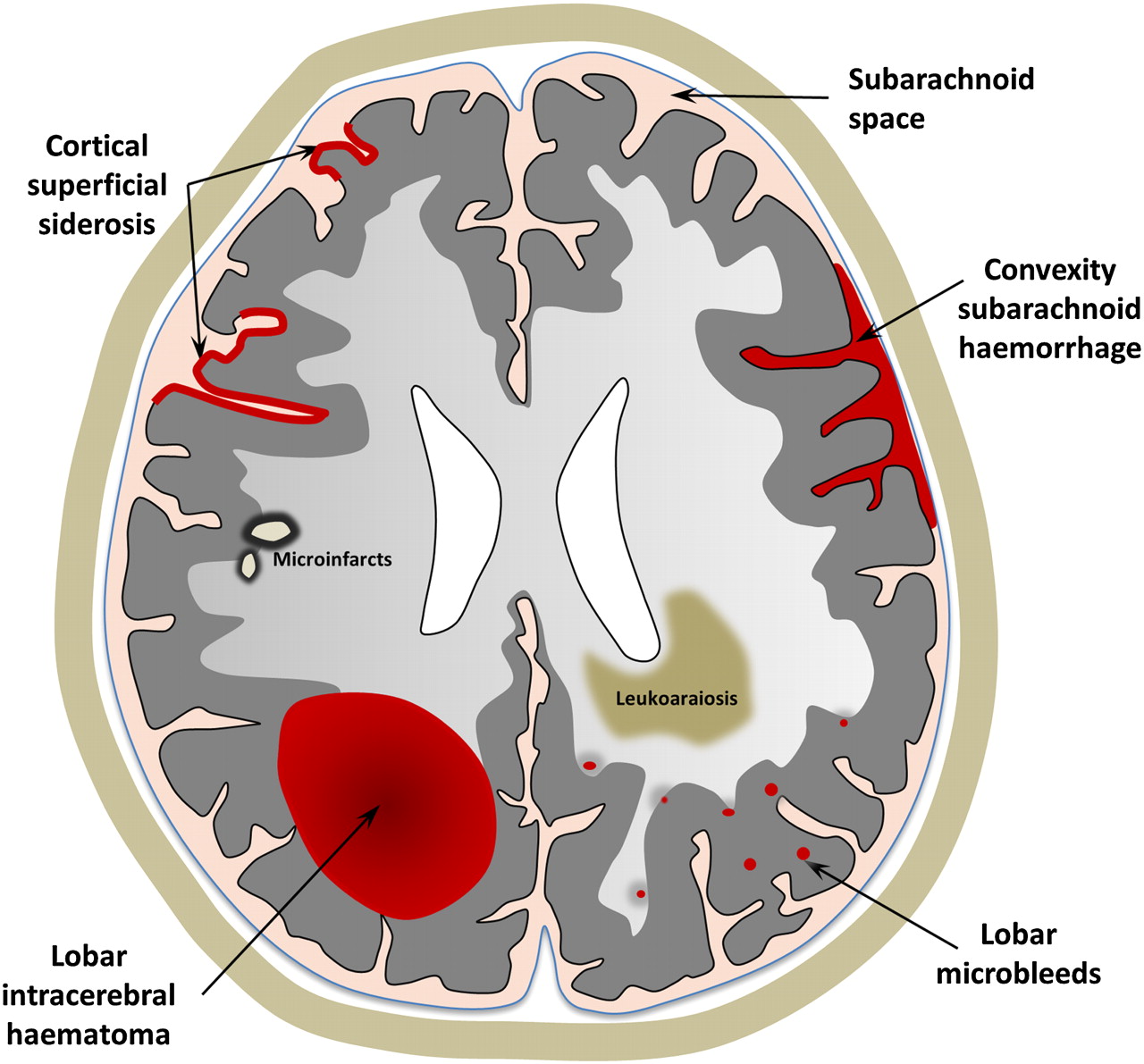
A schematic representation of the spectrum of haemorrhagic and ischaemic manifestations of sporadic cerebral amyloid angiopathy, visible on MRI.
Cerebral microbleeds
The widespread use of T2* weighted MRI sequences in the past decade or so has led to the increasing detection of CMBs: small, well demarcated, hypointense, rounded lesions, not detected on conventional MRI (figure 4D–G).136 Histopathological studies show that CMBs correspond to focal accumulations of haemosiderin laded macrophages (a blood breakdown product) adjacent to abnormal small vessels affected by hypertensive angiopathy or CAA.137 138 There is increasing evidence that hypertensive vasculopathy is associated with CMBs in deep brain regions (basal ganglia, thalamus and brainstem) whereas CAA is characterised by CMBs in a lobar distribution136 138 with a predilection for the parietal lobes.104 The Rotterdam scan study139 140 showed a strong association between strictly lobar (but not deep) CMBs and ApoE ɛ4, consistent with the well known relation of this allele with CAA.49 A recent imaging study in clinically probable CAA, using non-invasive amyloid imaging with Pittsburgh Compound B (PiB), found that CMBs correspond to areas with a high concentration of amyloid.141 Moreover, CMBs correlate with the risk of lobar ICH recurrence,142 suggesting an important role in prognosis (as well as diagnosis) in CAA.11
Recent population based studies have revealed a high percentage of community dwelling elderly people with strictly lobar microbleeds (particularly in the posterior brain regions), suggesting subclinical CAA.139 140 143 144 This may have important implications: if strictly lobar CMBs are validated as a diagnostic marker of CAA, such asymptomatic individuals could benefit from new therapeutic agents to reduce the progression of the disease.
Neuroimaging studies have revealed lobar CMBs in more than 20% of patients with AD,145 probably reflecting advanced CAA (in keeping with neuropathological findings). Patients with autosomal dominant forms of familial AD (who have a younger age at symptom onset), also seem to have a prevalence of lobar CMBs similar to sporadic AD146—a striking recent observation since these patients are much more likely to have ‘pure’ neurodegenerative AD without coexisting sporadic small vessel disease. It has been suggested that the presence of multiple lobar CMBs in patients with AD may identify a specific subgroup of patients with a different clinical phenotype with therapeutic implications which need to be explored in future studies.145
Leukoaraiosis
Leukoaraiosis is a radiological term which describes imaging changes (often confluent) in deep cerebral white matter. Leukoaraiosis appears as low attenuation on CT scans or hyperintensity on T2 weighted or FLAIR MRI, typically sparing subcortical U fibres (figure 4C).147 Pathological substrates include demyelination, axon loss and mild gliosis. The pathogenesis of leukoaraiosis in CAA probably involves chronic hypoperfusion of the vulnerable periventricular white matter and disruption of the blood–brain barrier due to amyloid in the overlying cortical small vessels.32 95 148 Another possible mechanism of leukoaraiosis in CAA is as a result of the accumulation of silent ischaemic lesions (microinfarcts).12 35 149 Indeed, leukoaraiosis is very common in CAA, preferentially involving posterior regions,150 although some studies suggest no major difference in the topography of leukoaraiosis in CAA compared with hypertensive arteriopathy.122 150 A recent investigation suggested that subjects with CAA related lobar ICH have a higher prevalence of occipital dominant leukoaraiosis compared with normal elderly controls151; this interesting finding requires further clinical attention and investigation. Leukoaraiosis may be an important contributor to overall disease burden, especially progressive cognitive impairment,152 given its tendency to accumulate over time.153 A recent study found that leukoaraiosis volume was greater in patients with CAA and hypertension than those without, suggesting that strict control of hypertension might reduce leukoaraiosis related disability in CAA.152
Convexity subarachnoid haemorrhage and cortical superficial siderosis
Atraumatic cSAH and superficial siderosis are recently recognised imaging correlates of sporadic CAA154 which seem to be quite characteristic of the disorder (figure 4E,F). cSAH is localised bleeding, usually in up to several adjacent sulci, without other subarachnoid bleeding at the base of the brain in the pattern typically associated with saccular aneurysm rupture.155 Although rare in isolation, in CAA, cSAH often results from lobar ICH extending to the cortical surface.134 154 156 The largest cohort of patients with isolated cSAH published (n=29) found that CAA was a frequent apparent cause in patients over 60 years old.155 157 A recent retrospective analysis of consecutive patients admitted to a tertiary stroke unit with cSAH suggested that CAA could be a common cause in the elderly, with a characteristic clinical presentation of single or recurrent transient focal neurological attacks.135 Another recent study of a cohort of patients presenting with cSAH reported similar findings.158
Cortical superficial siderosis describes haemosiderin deposition in the superficial layers of the cerebral cortex (figure 5), and may follow repeated episodes of bleeding in the subarachnoid space.154 On T2*-GRE MRI sequences, cortical superficial siderosis shows a characteristic ‘gyriform’ pattern of hypointense signal (figure 5E,F).154 Linn et al have recently detected cortical superficial siderosis in 47.4% (n=38) of patients with a clinical diagnosis of CAA compared with no controls (mean age 54 years), suggesting that it might be helpful for the clinical diagnosis of CAA (see below).159 Compared with the well known syndrome of CNS superficial siderosis, which typically affects the brainstem and posterior fossa (associated with cerebellar and brainstem signs), CAA related siderosis has a predilection for the cerebral convexity160 and may be associated with transient neurological manifestations.132
Silent acute ischaemic lesions on diffusion weighted imaging
Neuropathological evidence of asymptomatic ischaemic infarction is an established finding in the brains of patients with advanced CAA.8 67–69 Recent studies using magnetic resonance diffusion weighted imaging—which is extremely sensitive to even small areas of acute ischaemia—have shed light on the dynamics of this phenomenon in vivo (figure 4H). A case report13 and a recent case control study12 found a high prevalence of diffusion weighted imaging positive lesions in patients with advanced CAA. These lesions were associated with CMB burden, suggesting shared pathophysiological pathways.161 Gregoire et al recently established that acute, subclinical ischaemic brain lesions are frequent after recent acute ICH, and are three times more common in CAA related ICH than other spontaneous bleeds35; the lesions were associated with the severity of leukoaraiosis and lobar CMBs, suggesting that they were due to a CAA related occlusive arteriopathy.35 These data suggest a dynamic interplay between the haemorrhagic (‘microbleeding’) and ischaemic (‘microinfarction’) components in CAA161 although the therapeutic implications and prognostic significance of these findings require further study.
Molecular imaging of vascular amyloid in vivo
MRI indirectly detects the consequences of CAA (eg, CMBs, cSAH and siderosis) rather than vascular amyloid itself. Consequently, a large proportion of ‘silent’ CAA may be as yet undetectable. Positron emission tomography methods allow the in vivo imaging of amyloid in the brain, using several radioligands, of which the most widely studied is 11C PiB.162 Ly et al demonstrated that CAA subjects had increased global PiB uptake relative to a healthy elderly control group, and found an occipital predominance of PiB retention in CAA compared with AD.16 PiB positron emission tomography might therefore ultimately detect CAA in vivo, even before it causes symptomatic ICH or the known radiological sequelae, including CMBs.14 15 141 163
Diagnostic approach to cerebral amyloid angiopathy: the critical role of neuroimaging
A common clinical scenario where sporadic CAA should be suspected is in elderly patients presenting with lobar ICH. The most commonly used criteria for CAA diagnosis are the Boston criteria (box 2).164 In the absence of direct neuropathological examination, CAA is diagnosed based on characteristic neuroimaging findings.164 The diagnosis of ‘probable CAA’ requires the following (box 2):
Age ≥55 years
Detection of multiple haemorrhagic cerebral lesions
Haemorrhages confined to cortical or cortical–subcortical (lobar) brain regions
Exclusion of secondary causes of ICH, such as arteriovenous malformations, head trauma, brain tumour, vasculitis and excessive anticoagulation.
Classic and modified Boston criteria for the diagnosis of cerebral amyloid angiopathy (CAA). (*Modifications compared with the classic Boston criteria based on Linn et al159164)
1. Definite CAA
Full postmortem examination demonstrating:
Lobar, cortical or cortical–subcortical haemorrhage
Severe CAA with vasculopathy
Absence of other diagnostic lesion
2. Probable CAA with supporting pathology
Clinical data and pathological tissue (evacuated haematoma or cortical biopsy) demonstrating:
Lobar, cortical or cortical–subcortical haemorrhage
Some degree of CAA in specimen
Absence of other diagnostic lesion
3. Probable CAA
Clinical data and MRI or CT demonstrating:
Multiple haemorrhages restricted to lobar, cortical or cortical–subcortical regions (cerebellar haemorrhage allowed)
*[OR single lobar, cortical or cortical–subcortical haemorrhage and focalb or disseminatedc superficial siderosis]
Age ≥55 years
Absence of other cause of haemorrhagea
4. Possible CAA
Clinical data and MRI or CT demonstrating:
Single lobar, cortical or cortical-subcortical haemorrhage
*[OR focalb or disseminatedc superficial siderosis]
Age ≥55 years
Absence of other cause of haemorrhage1
aOther causes of haemorrhage (differential diagnosis of lobar haemorrhages):
Antecedent head trauma
Haemorrhagic transformation of an ischaemic stroke
Arteriovenous malformation
Haemorrhagic tumour
Warfarin therapy with international normalisation ratio >3
Vasculitis
bFocal siderosis: siderosis restricted to 3 or fewer sulci
cDisseminated siderosis: siderosis affecting at least 4 sulci
The specificity of the Boston criteria has been validated against the established gold standard of neuropathological diagnosis from autopsy, haematoma evacuation or cortical biopsy.164 In this study, the criteria showed excellent specificity: all cases identified as ‘probable CAA’ (n=13) had pathological evidence of severe CAA. However, the sensitivity of the probable category was 44%, so that it failed to identify over 50% of those with severe CAA pathology.164 However, this study does not reflect current radiological practice as only 15 patients had T2*-GRE imaging. Recently, the application of the Boston criteria with a greater use of T2*-GRE MRI in Dutch-type hereditary CAA found a much improved sensitivity (especially when lobar CMBs were included in the criteria).165 The rationale for the inclusion of lobar CMBs in the criteria is that both lobar CMBs and lobar ICH represent independent vascular rupture events which are considered to offer equal evidence for the presence of CAA.166 The recently introduced SWI, a three-dimensional T2*-GRE technique, enables visualisation of CMBs with much increased sensitivity, resulting in higher lesion counts (at least 67% more compared with conventional T2*-GRE) (figure 4D),167–169 but its effect on diagnostic accuracy for CAA requires further study. Superficial siderosis and cSAH, which have a high prevalence in CAA related ICH but are rare in other forms of ICH, have been shown to enhance the sensitivity of the Boston criteria without loss of specificity.159
Although the value of T2* weighted MRI and SWI in detecting CMBs, cSAH and siderosis has mainly been validated in cohorts of patients who presented with symptomatic ICH, such imaging may also have a role in the diagnosis of patients presenting without major haemorrhage but with other syndromes, raising suspicion of CAA; for example, elderly patients with progressive cognitive impairment.118 135 168 170 171 In addition, although at present T2* MRI or SWI sequences are not part of the routine investigation of TIA-like attacks, there might be useful in patients with CAA related transient focal neurological episodes (‘amyloid spells’)—rather atypical of TIAs (figure 4G).132 135 However, current data are insufficient to make evidence based recommendations.
Other biomarkers might also prove useful in the non-invasive diagnosis of CAA, in particular the assessment of Aβ concentrations in CSF. Decreased levels of CSF Aβ42 but not Aβ40 are found in AD172 while it has been reported that both Aβ42 and Aβ40 concentrations are decreased in CAA, relative to control and AD patients.173 It has also been suggested that the combination of low Aβ42 with increased total τ levels in CSF, can discriminate CAA patients from normal controls with high accuracy.173 Another potentially promising marker of CAA might include retinal changes (microaneurysms and dot and blot haemorrhages174). A critical goal of all of these potential approaches is to reliably identify CAA at the early (asymptomatic) stages of the disease, to allow the best chance for disease modifying or preventive treatments to be effective.
Management and prospects for disease modification
Acute treatment
No treatment is specific for symptomatic management of CAA or CAA related ICH. As in all forms of spontaneous ICH, CAA related haematomas enlarge in the first few hours after onset, providing a potential target for treatment. One of the most promising available treatments in acute ICH is lowering blood pressure, which has been shown to reduce haematoma expansion in a randomised trial,175 presumably by reducing hydrostatic pressure into the ICH in the critical hyperacute phase; a further large study is underway.176 The role of neurosurgery in ICH remains to be defined clearly and is a topic of ongoing investigation.177 Although there have previously been concerns regarding surgery in CAA due to the risk of bleeding from fragile amyloid laden vessels, the available evidence does not suggest a particularly high operative risk.178–180 Neurosurgery for haematoma evacuation appears relatively safe in at least some patients with CAA related ICH, particularly in patients less than 75 years of age without intraventricular extension.180 Until further evidence of specific acute treatments is available, it is reasonable to follow the American Heart Association Stroke Council guidelines for acute management of ICH, without modification for individuals with suspected CAA.181 Active research into new approaches for acute ICH treatment is expected to benefit patients with CAA related bleeds. Novel approaches, including neuroprotective drugs182 183 which target the multitude of processes that occur after ICH (eg, cerebral oedema, thrombin release, red blood cell lysis and haemoglobin induced neurotoxicity)184 and iron chelating agents (such as deferoxamine)185 are all being studied in early phase trials.9 181 186
Prevention of recurrent ICH
Withholding anticoagulants and antiplatelets
It is a paradox that many elderly patients at highest risk of occlusive vascular events are also at the highest risk of haemorrhage complications, including ICH. Judging the balance of risk and benefit of antithrombotic treatment after ICH in those patients with an indication for vascular secondary prevention is thus a major clinical challenge. The available evidence on this topic is limited, consisting of generally small case control and prospective observational studies. In a recent prospective cohort of patients with spontaneous lobar ICH, an association was found between aspirin use and ICH recurrence after adjusting for other potential ICH risk factors (HR 3.95, 95% CI 1.6 to 8.3; p <0.021).187 Rebleeding risk was associated with the number of lobar CMBs and the presence of leukoaraiosis in posterior brain regions—possible markers of underlying CAA and its severity.187 Gregoire et al in a small case control study found that lobar CMBs were associated with antiplatelet related ICH, also supporting a link between CAA and antiplatelet related ICH.188 Another small case control study of warfarin related ICH and matched ICH-free warfarin users showed an association of warfarin with CMBs, but with large CIs around the ORs for the association.189 There are no randomised trial data but a decision analysis suggested that in patients with CAA related ICH, the use of anticoagulants to prevent future cardioembolic (atrial fibrillation related) stroke would lead to an ICH rate that outweighs any benefit from the treatment.190
Whether multiple lobar CMBs (without symptomatic ICH) confer an unacceptably high risk of future ICH with the use of antithrombotic agents requires further study. In a recent meta-analysis, Lovelock et al pooled the information of 1461 patients with ICH and 3817 patients with ischaemic stroke or TIAs and they showed that CMBs were more common in warfarin related ICH than ‘spontaneous’ ICH. In pooled follow-up data for 768 patients treated with antithrombotics (anticoagulant or antiplatelet drugs), the presence of CMB at baseline was associated with a significantly increased risk of future ICH (OR 12.1; 95% CI 3.4 to 42.5; p<0.001), but this study did not separately investigate the effects of lobar versus deep CMBs.191
For the moment, anticoagulation should usually be avoided in patients with a diagnosis of CAA and symptomatic lobar ICH, unless there is a very compelling need to treat that could outweigh the very high risk of recurrent ICH (eg, life threatening pulmonary embolism or a mechanical heart valve). Although antiplatelet drugs probably also increase future ICH risk in CAA, it may be reasonable to consider them in selected patients with CAA for secondary prevention in whom the risk of intracerebral bleeding is judged to be low and the risk of occlusive vascular events high, based on their clinical and imaging characteristics. In primary prevention, the risk/benefit ratio may favour withholding treatment in patients with multiple lobar CMBs. Further randomised clinical trials are urgently needed to help clarify the optimum antithrombotic treatment in these different CAA patient groups.
Blood pressure control
A recent subgroup analysis of the PROGRESS trial reported that lowering blood pressure with the antihypertensive drug perindopril (with or without indapamide) reduced the risk of probable CAA related ICH by 77% (95% CI 19% to 93%) over a follow-up period of 3.9 years.38 Despite a small total number of CAA related ICH events, this is the first trial to show that blood pressure lowering treatment protects against CAA related ICH, regardless of the presence of hypertension.38 Blood pressure lowering may also be associated with a more general benefit in cardiovascular risk and mortality in patients over the age of 80 years.192 Thus most patients with CAA and a history of symptomatic ICH should be offered antihypertensive treatment.
Statins
Recently, concerns have been raised over statins as a risk factor for ICH, in light of the results of the SPARCL trial of atorvastatin in patients with stroke, which showed a small increase in the incidence of ICH among patients receiving high doses of the drug193; the hazard was higher for patients with baseline haemorrhagic compared with ischaemic stroke (HR 4.1 vs 1.6).194 A decision analysis showed that the risk of statin therapy likely outweighs any potential benefit in patients with recent lobar ICH.195 Thus although there are inadequate data for clear recommendations on statin use,181 they should perhaps be avoided in the setting of a recent CAA related ICH.196 For individuals with suspected CAA based on the presence of multiple lobar CMBs (without any associated macrobleeding) the risks and benefit of statin therapy are uncertain.11
Disease modifying agents
An important hope for the future treatment of CAA is to identify patients early in the natural history of the disease before ICH or dementia occurs, to allow the use of disease modifying therapies.197 Given the rarity of the inflammatory variant of CAA, it is unlikely that randomised data will become available to guide treatment, and it therefore seems reasonable to employ anti-inflammatory and immune modulating agents.197 198 However, future treatments for the great majority of sporadic CAA cases are likely to focus on preventing CAA progression by decreasing the production, deposition, toxicity and/or clearance of vascular amyloid. A candidate agent which might delay or inhibit the progression of CAA is tramiprosate, an ionic compound which binds soluble Aβ and interferes with the amyloid cascade.199 Tramiprosate has been shown to be a safe treatment option for patients with suspected CAA in a phase 2 study, supporting future efficacy trials.200 Emerging data from the use of secretases inhibitors and/or immunisation against Aβ in AD will be invaluable in guiding further efforts for disease modification in CAA.
Conclusions
During the past decade, there have been tremendous advancements in our understanding of CAA, relating to its pathophysiology, clinical spectrum, imaging manifestations and diagnosis.
Sporadic CAA is a common disease of the elderly and will become an increasingly important healthcare challenge as populations age further.
Sporadic CAA is an important contributor to cognitive decline and spontaneous or anticoagulant related lobar ICH.115
Transient neurological spells in CAA may be misdiagnosed as TIAs, but seem to have characteristic clinical features; they need to be recognised as treating them with antithrombotic drugs may increase the risk of future ICH.
Recent advances in neuroimaging have provided a new imaging window into the dynamic haemorrhagic and ischaemic features of CAA.
Lobar CMBs, cSAH and cortical focal superficial siderosis show promise to reliably diagnose CAA in life, although validation of these findings against their histopathological correlates requires further study.
Molecular imaging of Aβ may further improve our ability to detect this condition in vivo and define its true prevalence and burden.14–16 141
The rapidly developing field of transgenic mouse modelling has provided significant insights into the pathophysiology of human CAA, including the key pathogenetic role of the perivascular drainage pathway and the differential effects of different ApoE genotypes.3
Despite our improved understanding of CAA, there are still many questions to be answered in order to identify targets for therapeutic and preventive interventions. Exciting diagnostic and therapeutic developments are on the horizon for this fascinating small vessel disorder.
Acknowledgments
The authors thank Professor Sebastian Brander for providing some of the pathological images and Dr Estelle Healy for help in describing the histological slides. The authors are also most grateful to Dr Rolf H Jäger, Reader in Neuroradiology and Consultant Neuroradiologist at Queen Square, for assistance with MRI interpretation.
References
Supplementary materials
David Werring discusses the paper in the JNNP podcast
Footnotes
Funding AC receives research support from the Greek State Scholarship Foundation. DJW receives research support from the Department of Health/Higher Education Funding Council for England (Clinical Senior Lectureship Award) and the Stroke Association. This work was undertaken at UCLH/UCL who received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme.
Competing interests None.
Provenance and peer review Commissioned; externally peer reviewed.