Article Text

Abstract
Objective Autoantibodies against paranodal proteins have been described in patients with inflammatory neuropathies, but their association with pathology of nodes of Ranvier is unclear. We describe the clinical phenotype and histopathological changes of paranodal architecture of patients with autoantibodies against contactin-1, identified from a cohort with chronic inflammatory demyelinating polyradiculoneuropathy (n=53) and Guillain-Barré syndrome (n=21).
Methods We used ELISA to detect autoantibodies against contactin-1. Specificity of the autoantibodies was confirmed by immunoblot assay, binding to contactin-1-transfected human embryonic kidney cells, binding to paranodes of murine teased fibres and preabsorption experiments. Paranodal pathology was investigated by immunofluorescence labelling of dermal myelinated fibres.
Results High reactivity to contactin-1 by ELISA was found in four patients with chronic inflammatory demyelinating polyradiculoneuropathy and in none of the patients with Guillain-Barré syndrome, which was confirmed by cell binding assays in all four patients. The four patients presented with a typical clinical picture, namely acute onset of disease and severe motor symptoms, with three patients manifesting action tremor. Immunofluorescence-labelling of paranodal proteins of dermal myelinated fibres revealed disruption of paranodal architecture. Semithin sections showed axonal damage but no classical signs of demyelination.
Interpretation We conclude that anti-contactin-1-related neuropathy constitutes a presumably autoantibody-mediated form of inflammatory neuropathy with distinct clinical symptoms and disruption of paranodal architecture as a pathological correlate. Anti-contactin-1-associated neuropathy does not meet morphological criteria of demyelinating neuropathy and therefore, might rather be termed a ‘paranodopathy’ rather than a subtype of demyelinating inflammatory neuropathy.
- PERIPHERAL NEUROPATHOLOGY
- NEUROPATHY
- NEUROMUSCULAR
- NEUROIMMUNOLOGY
- GUILLAIN-BARRE SYNDROME
Statistics from Altmetric.com
Introduction
Autoimmune-mediated neuropathies comprise a pathogenically heterogeneous group of often disabling conditions. Autoantibodies against gangliosides can be found in a subset of patients with Guillain-Barré syndrome (GBS) and chronic inflammatory demyelinating polyneuropathy (CIDP), and some autoantibodies like GQ1b or diasilosyl antibodies are associated with distinct clinical syndromes.1 ,2 Nodes of Ranvier are suspected to be an important site of attack in immune-mediated neuropathies. Recently, autoantibodies directed against nodal and paranodal proteins have been detected in patients with CIDP and GBS,3–7 and disruption of nodal architecture has been described in patients with demyelinating neuropathy.8–10 The term ‘paranodopathy/nodopathy’ was, therefore, suggested to classify disorders with autoantibody-mediated damage to the paranodal/nodal region, which may represent a distinct pathogenic entity in a subset of patients with immune-mediated neuropathies not meeting the classical definition of a primary axonal or demyelinating lesion.11 ,12 CIDP with anti-neurofascin IgG4 is characterised by disabling tremor and poor response to intravenous immunoglobulin (IVIG).6 Contactin-1 is another paranodal protein that seems to induce an immune response. IgG4 antibodies against contactin-1 were detected in three patients with severe CIDP and prevented adhesive interaction of contactin-1/caspr complex and neurofascin 155 in cell culture.5 ,13 We tested for anti-contactin-1 antibodies in a cohort of patients with acute and chronic autoimmune neuropathy, studied the morphology of peripheral nerves in these patients and show that autoantibodies against contactin-1 affect nodal architecture. We present the first combined clinical and morphological characterisation of contactin-1 autoantibody-mediated inflammatory neuropathy.
Patients and methods
Patients
A cohort of 53 patients with CIDP (fulfilling Inflammatory Neuropathy Cause and Treatment (INCAT) criteria14), and 21 patients with GBS (fulfilling Brighton criteria15) were included. Among these, sera of 33 patients with CIDP and 8 patients with GBS were prospectively recruited in 2013 and 2014. Plasma exchange (PE) material of 33 patients (13 with GBS, 20 with CIDP) obtained between 2007 and 2012, and stored at −80°C in our department was retrospectively included. Sixty sera of healthy individuals and 10 sera of patients with other autoimmune diseases (myasthenia gravis, multiple sclerosis) served as controls. All participants gave informed consent to take part in the study. Clinical data and nerve conduction studies (NCS) were obtained from the patients’ records. Demographic data are summarised in table 1.
Demographic data of patients and controls
ELISA
Maxisorb 96-well plates (Thermo Fisher Scientific, Waltham, Massachusetts, USA) were coated with human contactin-1 (Sino Biological, Beijing, China) and incubated with patients’ samples (1:100) or anti-contactin-1 (mouse monoclonal, Abcam, Cambridge, UK, 1:200) and appropriate peroxidase-conjugated secondary antibodies (anti-human IgG or IgM for patients’ material, 1:10 000 /1:7000, DakoCytomation, Glostrup, Denmark; 1:500 anti-mouse IgG for the control antibody). Optical density was measured at 450 nm with Multiscan EX Elisa Reader (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Normal controls were run with each assay to determine the threshold value, which was set at five SDs above the mean of all normal controls (figure 1). To determine the IgG subclass, peroxidase-conjugated antihuman IgG1, IgG2, IgG3 and IgG4 secondary antibodies were used (Life Technologies, Carlsbad, California, USA; 1:1000 IgG1, 1:4000 IgG2, 1:3000 IgG3, 1:2000 IgG4). Titres of all positive patients were determined by diluting samples from 1:100 to 1:50 000. IgG levels were measured by human IgG ELISA kit (AssayPro, St Charles, Missouri, USA) according to the instructions of the manufacturer.
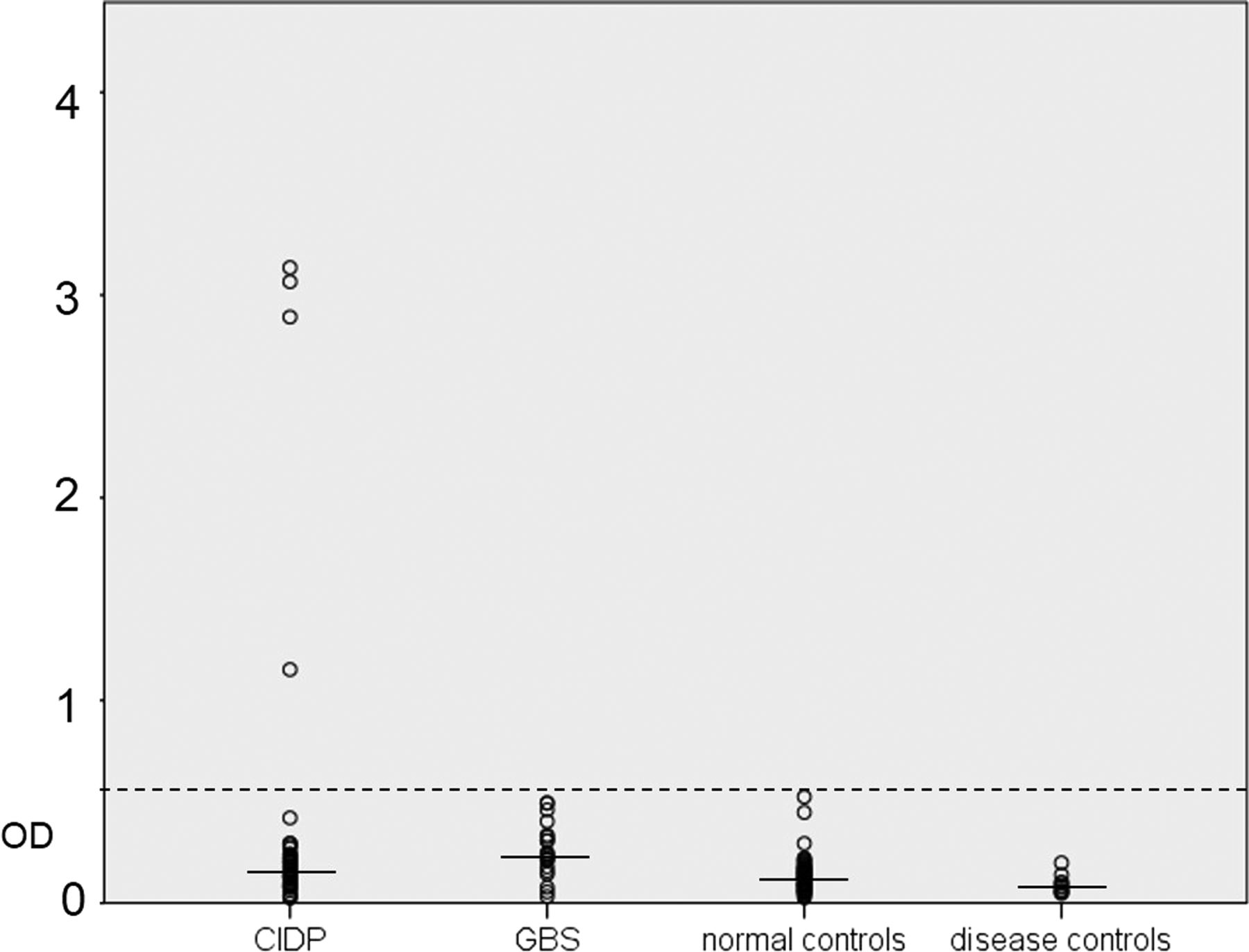
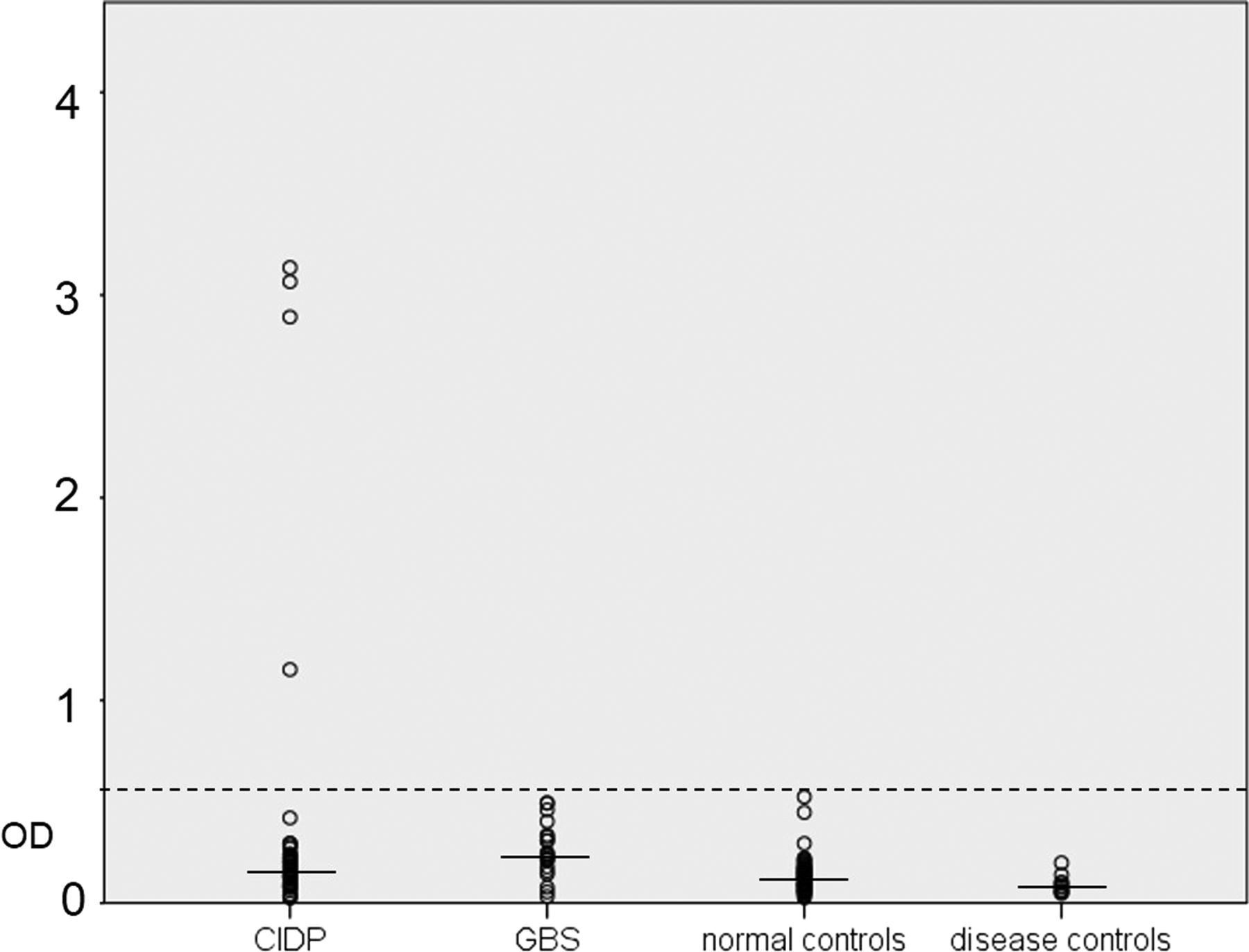
Optical density of samples from patients with CIDP and GBS, normal and disease controls reacted by ELISA. Four patients with CIDP showed reactivity above the normal range (dashed line, five SDs above the mean of normal controls; solid lines mark medians). CIDP, chronic inflammatory demyelinating polyneuropathy; GBS, Guillain-Barré syndrome; OD, optical density.
Binding assays with teased fibres and transfected human embryonic kidney 293 cells
Teased fibre preparations of murine sciatic nerves were incubated with patients’ sera or PE material, each at two different dilutions (1:100 and 1:500) and with appropriate anti-human Cy3-conjugated secondary antibodies (Dianova, Hamburg, Germany, 1:100). Double-immunofluorescence with anti-caspr (Abcam, 1:1000) was performed in positive cases to evaluate colocalisation at the paranodes. Slides were assessed using a fluorescence microscope (Zeiss Ax10, Zeiss, Oberkochen, Germany).
Human embryonic kidney (HEK) 293 cells were transiently transfected with plasmids of rat contactin-116 using the calcium-phosphate precipitation method.17 Cells were fixed with 4% paraformaldehyde, and were incubated with patients’ sera and PE material (1:500) and anti-contactin-1 (Abcam, 1:200) as a control antibody, and appropriate secondary antibodies (Cy3 and AlexaFluor488-conjugated anti-human IgG or anti-mouse IgG); these were assessed using a fluorescence microscope (Zeiss Ax10, see above).
Western blot
Contactin-1 protein (Sino Biological) was loaded onto polyacrylamide gels for electrophoresis and separated proteins were blotted onto nitrocellulose membranes, and were incubated with sera and PE material of different patients (1:2000) and peroxidase-conjugated anti-human IgG (Dako, Glostrup, Denmark). Reactive proteins were detected using ECL Plus Detection Kit (Perkin Elmer, Waltham, Massachusetts, USA).
Preabsorption experiments
Preabsorption was performed by incubation of patients’ samples (1:500 and 1:1000) with contactin-1-transfected HEK293 cells at room temperature. Sera incubated in wells with green fluorescent protein (GFP)-transfected cells served as controls. After incubation for 3 h, sera were used for binding assays on teased fibres as described above.
Binding assays with murine kidney and cerebellum sections
Immunofluorescence staining of mouse kidney sections was performed by incubation with patient and control samples, each at dilutions of 1:100 and 1:500, and consequent incubation with anti-human Cy3-conjugated secondary anti bodies (Dianova, 1:100). Immunostaining of rat cerebellum with patients’ and control sera was performed with a dilution of 1:100 and 1:500 followed by incubation with anti-human IgG (Dako, Glostrup, Denmark) and visualised with diaminobenzidine.
Skin and nerve biopsies
Punch skin biopsies of the distal and proximal leg of patient 3 were taken in 2008 and 2014. Skin biopsy of patient 4 was taken from the distal leg in 2007. For comparison, skin biopsies of 10 healthy controls, 14 of the patients with anti-contactin-1-negative CIDP and 3 patients with GBS were analysed. Double-immunofluorescence with anti-myelin basic protein (MBP) (Ultraclone, Isle of Wight, UK, 1:800), anti-caspr (Abcam, 1:100) and anti-pan-neurofascin (Abcam, 1:400) was performed as previously described.18 Intraepidermal nerve fibre density was determined by immunolabelling with anti-PGP9.5 (Ultraclone, 1:1000) and counting of nerve fibres crossing the dermal-intraepidermal junction according to published counting rules.19 All biopsies were evaluated using a fluorescence microscope (Zeiss Ax10, see above). Nodal length was determined by measuring the nodal gap using ImageJ as previously described.18 Sural nerve biopsies were performed according to standard procedures in patients 2–4 and in 28 patients with CIDP without detection of anti-contactin-1.20
A more detailed description of the methods is given in the supplements (see online supplementary methods).
Results
Detection of anti-contactin-1 antibodies by ELISA
A total number of 74 patient samples (41 sera, 33 PE material; 53 CIDP, 21 GBS) were tested for autoantibodies against contactin-1 by ELISA. Total IgG levels of sera ranged from 4.62 to 31.5 mg/mL (mean 19.23, SD 6.59), and IgG levels of PE material ranged from 5.44 to 31.44 mg/mL (mean 11.09, SD 6.22). IgG levels of control sera ranged from 9.3 to 28.76 mg/mL (mean 15.21, SD 4.89). Strong reactivity against contactin-1 in ELISA assays using human contactin-1 protein was found in the IgG fraction in four patients with CIDP (figure 1). We did not detect any IgM contactin-1 autoantibodies. Titres of IgG anti-contactin-1 were determined by dilution of all four positive samples from 1:100 to 1:50 000, and ranged from 1:10 000 (serum of patient 3) to 1:200 (patient 4; see online supplementary table). To determine the class of IgG, ELISA of all positive samples was repeated, using secondary antibodies directed against IgG1, IgG2, IgG3 and IgG4. Autoantibodies consisted of all four subtypes of IgG, but anti-IgG4 was the most abundant autoantibody found by ELISA in patients 2 and 3, and IgG3 was the most abundant in patients 1 and 4 (figure 2). Samples were obtained during the acute phase of the disease for patients 1 (3 months after onset) and 4 (1 month after onset), and during the chronic progressive phase of the disease for patients 2 (15 years after onset) and 3 (8 years after onset).

Reactivity to contactin-1 by patients’ IgG1–4 (y-axis: optical density). In patients 1 and 4, IgG3 is the predominant IgG subclass, in patients 2 and 3 IgG4 is predominant.
Confirmation of specificity of the contactin-1 autoantibodies
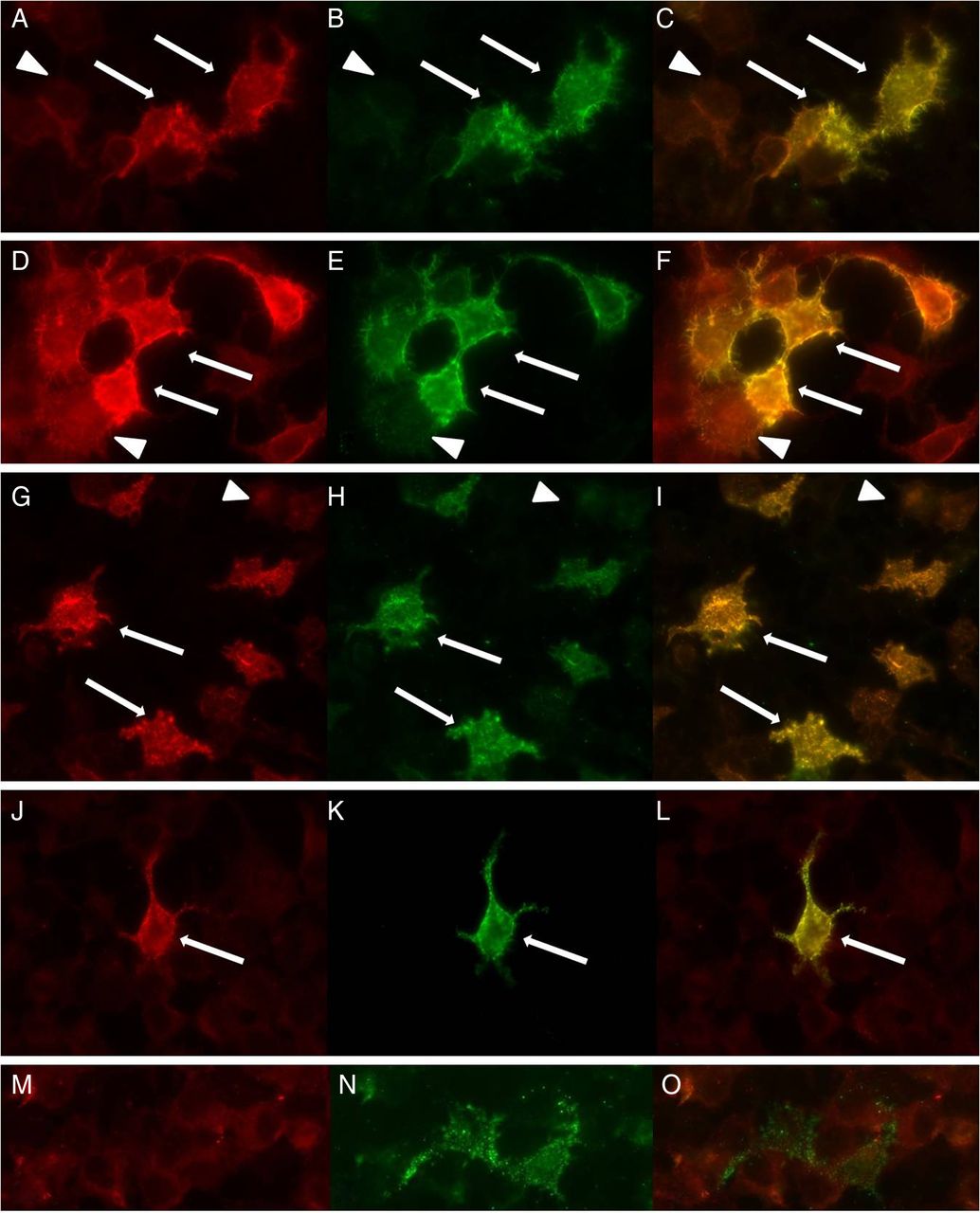
To assess specificity of the patients’ autoantibodies against contactin-1, contactin-1-transfected HEK293 cells were incubated with each serum/PE sample. Samples of all four patients with strong reactivity in ELISA showed clear binding to contactin-1-transfected cells but not to GFP-transfected control cells (figure 3). To confirm specificity on native tissue, binding assays using mouse teased nerve fibres and sera/PE samples from all patients and controls were performed. Distinct binding to the paranodes and mesaxon, and colocalisation with caspr were observed in experiments using sera from patients 1–3, but not from patient 4 or any other patient or control sample (figure 4). To further confirm specificity of the autoantibodies, western blot using human contactin-1 protein was performed with the sera samples of all four positive patients, five patients with GBS and six normal controls. Western blot showed clear reactive bands at approximately 150 kDa, identical to the band seen when staining with the control contactin-1 antibody (figure 5) in three of the patients with positive results in ELISA and HEK293 cells (patients 1, 3 and 4), but not in any other patient or control sample. Finally, preincubation of the sera of patient 1–3 with contactin-1-transfected HEK293 cells resulted in loss of paranodal staining of teased fibres (figure 6).

Double-immunofluorescence of contactin-1-transfected HEK293 cells with sera/PE material of patient 1 (A), patient 2 (D), patient 3 (G), patient 4 (J) and a normal control (M) and anti-contactin-1 (B, E, H, K, N). Clear colocalisation can be found in transfected cells (arrows) whereas untransfected cells (arrowheads) do not show immunolabelling with patients’ samples or anti-contactin-1. HEK, human embryonic kidney; PE, plasma exchange.
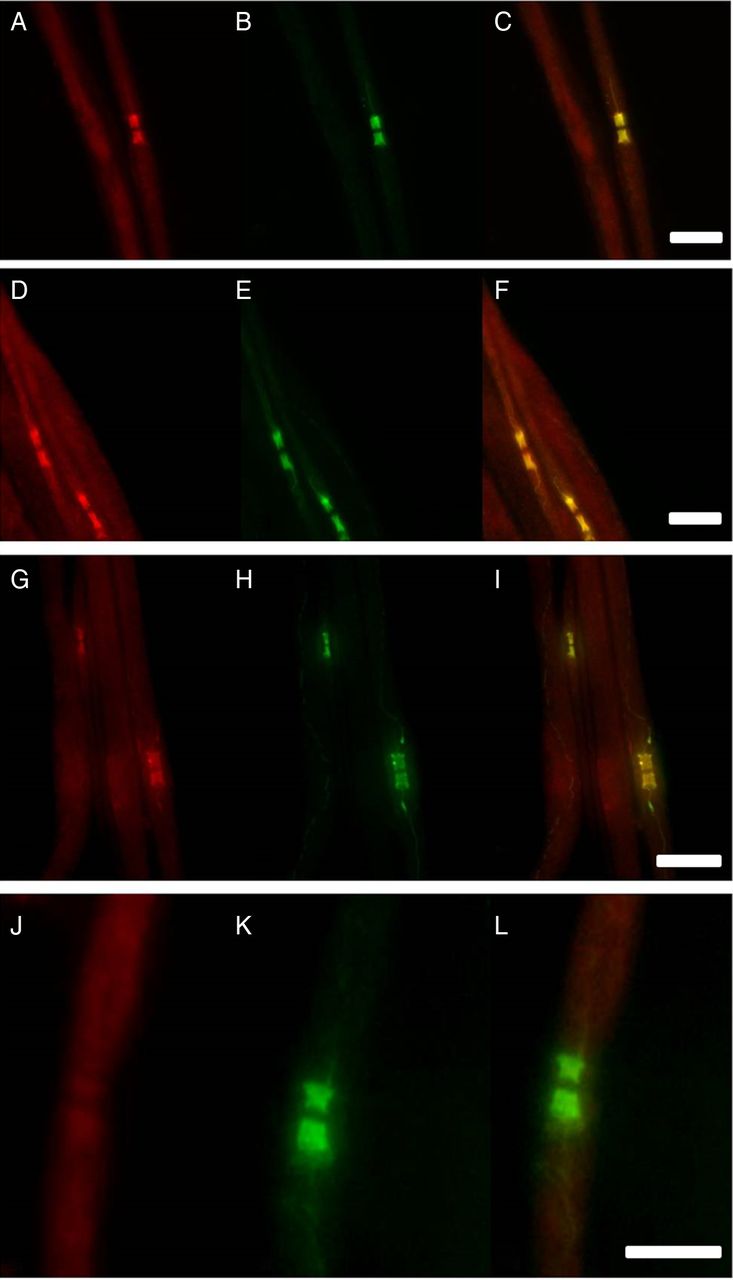
Double-immunofluorescence of murine teased fibres with samples of patient 1 (A), patient 2 (D) and patient 3 (G), a normal control (J) and anti-caspr (B, E, H, K). Overlay images (C, F, I, L) illustrate clear colocalisation of staining at the paranodes and mesaxon in patients 1–3 (A–I), not in the control (J–L). Scale bar=10 µm.

Western blot analysis of samples of patients 1–4, anti-contactin-1 and two normal controls. Clear protein bands near 150 kDa are marked by anti-contactin-1 and samples of patients 1, 3 and 4, not by the control samples and patient 2. In patient 1, purified IgG was tested to confirm the specificity of binding (PE, plasma exchange).

Preincubation of patient sera with contactin-1-transfected HEK293 cells and subsequent immunoreaction of murine teased fibres resulted in complete loss of paranodal staining (B) while immunoreactivity to paranodes was preserved after preincubation with GFP-transfected HEK293 cells (A) (patient 3, dilution of 1:500). Arrows mark nodal gaps. Scale bars=10 µm (HEK, human embryonic kidney).
Clinical features of neuropathy with anti-contactin-1 antibodies
Clinical data of all four patients with strong reactivity by ELISA are summarised in the online supplementary table. Remarkably, all patients had initially been diagnosed as GBS because of acute and rapidly progressive distal and proximal weakness and distal sensory symptoms, but diagnosis was modified to CIDP due to progression of symptoms after a short period of initial improvement.
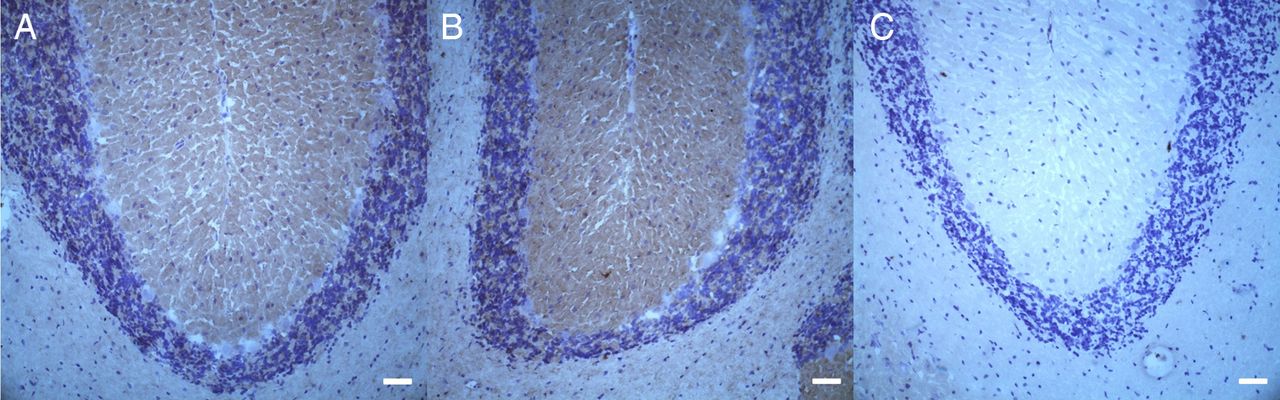
Patient 1 had an acute onset of disease and improved on treatment with IVIG. However, a few weeks later, he developed severe progressive sensorimotor neuropathy, which responded partially to plasmapheresis. Unfortunately he died due to sepsis, 4 months after onset of disease in 2012. Patient 2 experienced acute onset of neuropathy in 1991 and was admitted to our hospital in 2012. Records from other hospitals described an initial improvement to treatment with IVIG after acute and severe onset of disease, but subsequent progressive sensorimotor neuropathy with alternating episodes of remission and relapse. Patient 3 was first diagnosed in 2006 and regularly attended our clinic for diagnostic work-up and treatment since 2008. Clinical course was similar to patient 2, with acute onset of disease and progression of neuropathy interspersed with episodes of stabilisation. In patient 4, disease started in 2006 with acute onset. He initially improved with IVIG treatment, but then developed progressive peripheral neuropathy with episodes of remissions and relapses. He achieved almost normal muscle strength after a 6-month course of cyclophosphamide. He last attended our clinic in 2009 and was lost to follow-up due to dementia. According to his wife, muscle strength has remained stable since then. In summary, all patients presented with acute onset of motor more than sensory neuropathy and subsequent relapsing-remitting course of severe sensorimotor neuropathy. Action tremor was found in patient 2; patients 3 and 4 had rest and action tremor. Tremor analysis was performed in patient 3 and revealed 3.7 Hz non-length-dependent presumably cerebellar tremor of high amplitude accompanied by an underlying 2 Hz paretic tremor (see online supplementary video). To investigate a possible direct binding of autoantibodies to neurons of the cerebellum, binding assays using rat cerebellum sections were performed. Sera of patients 1 and 3 showed reactivity to the molecular and granular layers, but not to Purkinje cells. There was no binding detected in patients 2 and 4 and a normal control (figure 7). NCS of all patients showed prolonged distal latencies, decrease of nerve conduction velocity and prolonged F wave latencies, which worsened during the course of disease. Total cerebrospinal fluid protein was elevated in all four patients (see online supplementary table).

Immunohistochemical binding of patient and control sera to rat cerebellum. Binding to the molecular and granular layer of cerebellar cortex was detectable in patient 1 (A) and 3 (B) compared with a normal control (C). Scale bars=50 µm.
Patient 2 had suffered from membranous glomerulonephritis with nephrotic syndrome at the onset of disease, but later recovered completely from renal dysfunction. To investigate possible binding of patient sera to the kidney as further evidence of a pathogenic link between autoantibodies and renal symptoms, binding assays with all four patients’ material on mouse kidney sections were performed, but did not show any increase of reactivity compared with normal controls (data not shown).
Having identified a clinical picture of acute, GBS-like onset with progression to CIDP in all four patients with reactivity to contactin-1 by ELISA, we reanalysed the entire cohort for this phenotype. The cohort comprised of three additional patients with acute GBS-like onset of CIDP, but in these we could not find reactivity to contactin-1. A tremor similar to the one seen in patients 2–4 was found in one patient with anti-contactin-1-negative CIDP.
Histological findings of anti-contactin-1-associated neuropathy
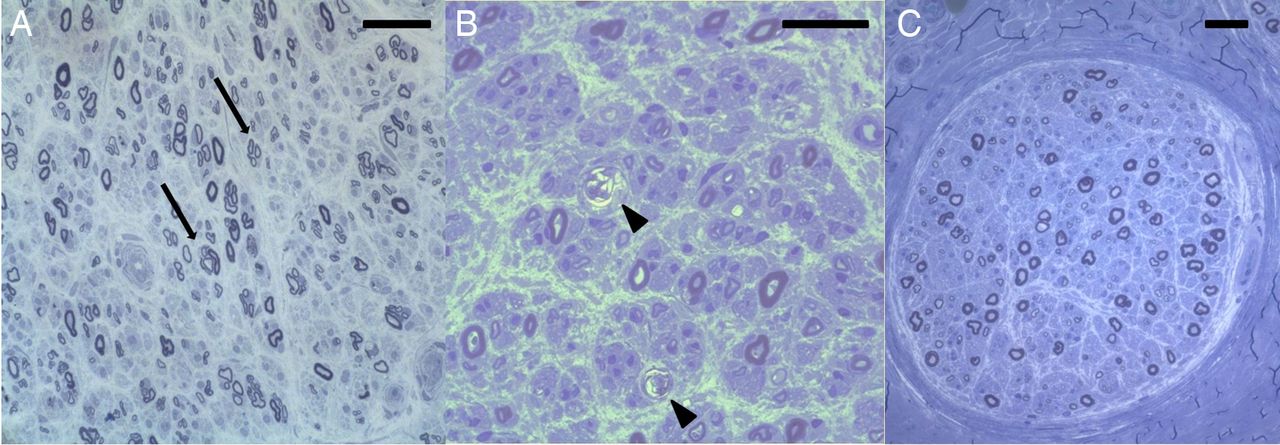
Sural nerve biopsy was performed in patients 2, 3 and 4. Numerous macrophages were present in the endoneurium whereas numbers of T cells were not elevated. Semithin sections showed axonal loss and degeneration of nerve fibres (figure 8). There were no onion bulbs and only few thinly myelinated fibres, resulting in a histopathological diagnosis of axonal neuropathy in all three patients. In contrast, only 2/28 sural nerve biopsies of patients with CIDP without anti-contactin-1 antibodies were classified as axonal; the others were categorised as demyelinating due to onion bulbs (13/28) and/or numerous thinly myelinated or naked fibres (23/28).

Semithin sections of sural nerve biopsies of patients 2–4 show clusters of regenerating nerve fibres (A, arrows, patient 2), degenerating fibres (B, arrowheads, patient 3) and axonal loss (C, patient 4). No onion bulbs and only few thinly myelinated fibres are visible. Scale bars=50 µm.
Analysis of myelinated fibres in skin biopsies from patient 3 (proximal leg, 2 years after onset of disease), and patient 4 (distal leg, 1 year after onset of disease) revealed elongated nodes of Ranvier (patient 3:7/10 nodes, patient 4:11/15 nodes) and loss/destruction of paranodal caspr and/or neurofascin immunoreactivity (caspr: patient 3:10/13 nodes, patient 4:12/15 nodes; neurofascin: patient 3:8/12 nodes, patient 4:10/14 nodes; figure 9). In comparison, 3 skin biopsies of patients with GBS and 14 biopsies of patients with anti-contactin-1-negative CIDP were analysed and revealed elongated nodes in 5/14 patients with CIDP (median: 2/6 nodes), but none in the patients with GBS. No loss/destruction of caspr/neurofascin immunoreactivity was found in any patient with GBS or anti-contactin-1-negative CIDP, but expansion of caspr and/or neurofascin staining to the juxtaparanodes, as previously described,8 was detected in 1/3 patients with GBS and in 7/14 patients with anti-contactin-1-negative CIDP. The mean nodal length in the two anti-contactin-positive patients was 7.1 (±1.8) µm, compared with our intralaboratory mean control value of nodal length of 4.3 (±0.9) µm.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Skin biopsy sections of patient 3 and 4 were immunolabelled with anti-MBP/anti-caspr (A–C, representative picture of patient 3) and anti-MBP/anti-neurofascin (D–F, representative picture of patient 4). Double-immunofluorescence of a dermal nerve bundle of patients 3 and 4 with anti-MBP (A, E) and anti-caspr (B) or anti-pan-neurofascin (F). Double-labelling (C, G) reveals complete loss of caspr at the paranodes (arrow) and severe disruption of caspr-staining and elongation of the nodal gap (arrow-head) and disrupted neurofascin staining (G). (D) and (H) show nodes of a normal control stained with anti-caspr (D) and anti-pan-neurofascin (H) with clear localisation of caspr at paranodes and neurofascin at nodes and paranodes. Scale bars=20 µm.
Another skin biopsy of the proximal leg and finger of patient 3, taken in 2014, showed complete depletion of myelinated fibres.
Follow-up after treatment with rituximab
Patient 3 was treated with rituximab in June 2014. Some weeks after therapy, her gait and tremor improved (see online supplementary video, taken after improvement of tremor and gait). At a follow-up in December 2014, proximal strength had returned to normal and distal strength had improved. The tremor had disappeared. The anti-contactin-1 titre measured by ELISA had decreased from 1:10 000 (April 2014) to 1:3000 (December 2014). The ratio of IgG subtypes had not relevantly changed.
Discussion
In the present study, we analysed paranodal architecture and histopathology of patients with autoantibodies against contactin-1. Biochemical and cell-based assays were used to identify patients with contactin-1 autoantibodies and specificity was further confirmed by binding of sera from three patients to paranodes of murine teased fibres and the loss of immunostaining at paranodes in autoantibodies depletion experiments using preabsorption to contactin-1-transfected HEK293 cells. Autoantibodies against contactin-1 have recently been identified by Querol et al5 using binding assays with transfected HEK293 cells. We further demonstrate functional effects of these autoantibodies by providing evidence for paranodal disruption in peripheral nerves of patients with contactin-1 autoantibodies as the presumed morphological correlate of autoantibody attack and give the first description of sural nerve biopsy findings of this entity that contrasts to the histological picture seen in classical CIDP. Our data result in the description of a uniform and detailed clinical and histological picture of patients with anti-contactin-1 autoantibodies, which is consistent with a paranodopathy classification.
All patients with contactin-1 autoantibodies presented with acute GBS-like onset, severe motor and moderate sensory symptoms and relapsing-remitting course of disease, resembling the clinical data given by Querol et al.5 In a letter by Miura et al,21 anti-contactin-1 was detected in none of 14 patients with acute-onset CIDP, but in 2 patients, each with CIDP and GBS. However, clinical data in this report are sparse and larger studies are needed to clearly define the clinical picture of anti-contactin-1-positive patients. In two patients (patients 1 and 4), we collected samples during the acute phase of the disease, and found that IgG3 was the most abundant subclass of anti-contactin-1 antibodies. Remarkably, anti-ganglioside autoantibodies in GBS and Miller-Fisher syndrome, another acute autoimmune neuropathy, are also of the IgG3 subclass.22–24 In the two patients that were studied during chronic progression of disease (patients 2 and 3), IgG4 was the most abundant subtype, as described in other cases of chronic inflammatory neuropathy with autoantibodies against contactin-1 and neurofascin-155.3 ,6 ,13 Switch of IgG subclasses has been described in membranous glomerulonephritis, with IgG1 at the onset of disease and IgG4 during the chronic stage,25 and in patients with multiple sclerosis treated with glatiramer acetate who produce IgG1 antibodies first and switch to the production of IgG4.26 A switch from IgG3 to IgG4 might occur in patients with acute-onset anti-contactin-1-associated neuropathy, but needs to be confirmed in longitudinal studies. The predominance of IgG3 at the onset of disease might explain the initial response to IVIG, while the switch to IgG4 might explain the lack of response at later stages, consistent with the view that patients with IgG4 autoantibodies are poor responders to treatment with IVIG as IgG4 does not induce complement activation.6
Both patients with predominant IgG4 anti-contactin-1 suffered from action tremor, resembling the clinical picture described in patients with IgG4 anti-neurofascin-155.6 As contactin-1 binds to neurofascin-155, the same site of attack underlies both subtypes of CIDP and we assume that acute-onset severe motor neuropathy rather than sensory neuropathy, often associated with action tremor, constitutes the typical clinical phenotype of IgG4-mediated paranodopathy encompassing anti-contactin-1 and anti-neurofascin-155 neuropathy. Complete absence of paranodal binding after preabsorption of the patients’ sera with contactin-1-transfected HEK293 cells strongly argues against additional neurofascin-155 antibodies in the patients described in our study. Binding assays of patients 1 and 3 argue for direct binding of anti-contactin-1 to cerebellar structures. However, patients 2 and 4 suffered from tremor but did not show any cerebellar binding. This might be due to the lower titres of patients 2 and 4 (1:1500; 1:200) compared with patients 1 and 3 (1:7400; 1:10 000).
Nephrotic syndrome occurred at the acute onset of disease in patient 2 who was diagnosed with membranous glomerulonephritis. Focal segmental and membranous glomerulonephritis have been described in a number of patients with severe CIDP and GBS.27–29 Autoantibodies could have contributed to the kidney deficits because contactin-1 is expressed in the kidney at low levels.30 However, incubation of samples with murine kidneys did not detect any binding, arguing against in situ formation of immune complexes and rather supports the alternative explanation of immune complex deposition as assumed in other forms of secondary membranous glomerulonephritis.
Nodal/paranodal damage caused by antiganglioside antibodies is supposed to be mediated by complement activation.31–33 However, IgG4, the most abundant subtype of autoantibody found in two of our patients during the chronic phase of disease, does not induce activation of complement. Damage of paranodal architecture by anti-contactin-1 in IgG4 paranodopathy, therefore, may be induced by direct binding of autoantibodies to the paranodes. Labasque et al13 have demonstrated that anti-contactin-1 antibodies of patients prevent adhesive interaction between contactin-1/caspr and neurofascin-155 and lead to alteration of paranodes in myelinated fibres in neuronal culture. Our findings provide the first in vivo description of nodal/paranodal architecture in peripheral nerves of patients with anti-contactin-1 neuropathy. Severe disruption of paranodal and nodal architecture with loss of caspr and/or neurofascin-immunoreactivity at some nodes and elongation of the nodal gap suggests an anti-contactin-1 pathogenic mechanism which leads to impaired nerve conduction in this disorder.
When introducing the term nodopathy/paranodopathy, Uncini et al12 referred to patients with antiganglioside antibodies and conduction block, but nearly normal nerve conduction velocities. In contrast, anti-contactin-associated neuropathy is associated with a reduction of nerve conduction velocity. Disruption of paranodes with loss of axoglial interacting proteins and elongation of the nodal gap, as shown in our patients, indicate that autoantibodies against contactin-1 could lead to changes of the paranodes that result in slow nerve conduction and/or conduction block. The observations of reduced nerve conduction velocities and increased distal latencies in our patients are, in fact, consistent with earlier biophysical studies that showed a dependence of conduction speed on preserved paranodal architecture.34 ,35 Semithin sections revealed axonal loss without typical features of demyelinating neuropathy, like numerous thinly myelinated fibres or onion bulbs, supporting the idea that damage to the paranodes rather than the internodal myelin sheath causes impairment of nerve conduction in this subtype of CIDP. We, therefore, suggest that anti-contactin-associated neuropathy represents another form of paranodopathy, resulting in the disruption of paranodal architecture, subsequent axonal loss and slower nerve conduction that do not follow the classical demyelinating neuropathies that are characterised by features of demyelination and remyelination leading to thinly myelinated fibres or onion bulbs.
We conclude that anti-contactin-1-mediated paranodopathy comprises a new subgroup of autoimmune peripheral neuro-pathy with a distinct clinical phenotype, induced by damage to the paranodes. Testing of anti-contactin-1 might be warranted in patients with acute-onset of severe sensorimotor peripheral neuropathy.
Acknowledgments
The authors thank Sonja Mildner, Susi Hellmig, Barbara Broll, Barbara Dekant and Nadine Vornberger for their expert technical assistance. Nerve biopsy sections of patient 2 have been kindly provided by Professor Bergmann, Department of Neuropathology, Klinikum Bremen-Mitte.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
- Data supplement 2 - Online table
- Data supplement 3 - Online video
Footnotes
Contributors KD was involved in conception, execution and organisation of the study, recruitment of patients, evaluation of data and statistical analysis, writing of manuscript. LA was involved in execution of experiments, evaluation of data, recruitment of patients. KW was involved in execution of experiments, evaluation of data. CV was involved in conception of the study. SDD-H and SGW provided critical reagent and edited manuscript. MM was involved in recruitment of patients. AW was involved in conception and execution of the study, execution of experiments, evaluation of data. CS was involved in conception and organisation of the study, recruitment of patients, evaluation of data, writing of manuscript.
Funding The study was supported by research funds of the University of Würzburg. Work in the laboratory of SDD-H and SGW was supported in part by grants from the Rehabilitation Research Service and Medical Research Service, Department of Veterans Affairs, USA. LA is supported by a grant of the Graduate School of Life Sciences of the University of Würzburg.
Competing interests CS has received personal fees for educational talks for Baxter, CSL Behring and Kedrion. KD has received personal fees for educational talks for Baxter. SGW serves as an advisor to SiteOne Pharmaceuticals and has received consulting fees from Janssen Pharmaceuticals and Chromocell Corporation. SDD-H has received personal fees from Ono Pharmaceuticals, Guidepoint Global and Endopharmaceuticals.
Patient consent Obtained.
Ethics approval Ethics committee of the Medical Faculty, University of Würzburg.
Provenance and peer review Not commissioned; externally peer reviewed.