

Abstract
Clustering of Na+ channels at the nodes of Ranvier is coordinated by myelinating glia. In the peripheral nervous system, axoglial contact at the nodes is mediated by the binding of gliomedin and glial NrCAM to axonal neurofascin 186 (NF186). This interaction is crucial for the initial clustering of Na+ channels at heminodes. As a result, it is not clear whether continued axon-glial contact at nodes of Ranvier is required to maintain these channels at the nodal axolemma. Here, we report that, in contrast to mice that lack either gliomedin or NrCAM, absence of both molecules (and hence the glial clustering signal) resulted in a gradual loss of Na+ channels and other axonal components from the nodes, the formation of binary nodes, and dysregulation of nodal gap length. Therefore, these mice exhibit neurological abnormalities and slower nerve conduction. Disintegration of the nodes occurred in an orderly manner, starting with the disappearance of neurofascin 186, followed by the loss of Na+ channels and ankyrin G, and then βIV spectrin, a sequence that reflects the assembly of nodes during development. Finally, the absence of gliomedin and NrCAM led to the invasion of the outermost layer of the Schwann cell membrane beyond the nodal area and the formation of paranodal-like junctions at the nodal gap. Our results reveal that axon-glial contact mediated by gliomedin, NrCAM, and NF186 not only plays a role in Na+ channel clustering during development, but also contributes to the long-term maintenance of Na+ channels at nodes of Ranvier.
Introduction
The nodes of Ranvier are short gaps between adjacent myelin segments characterized by the presence of a high local density of voltage-gated Na+ channels (Waxman and Ritchie, 1993). The concentration of these channels at the nodal axolemma allows rapid nerve impulse conduction along myelinated axons. At the nodes, Na+ and KCNQ2 K+ channels are found in a complex with the adaptor protein ankyrin G, the cytoskeletal protein βIV spectrin, and the Ig superfamily cell adhesion molecules NrCAM and neurofascin 186 (NF186; Poliak and Peles, 2003; Salzer, 2003). The latter functions in node formation by recruiting ion channels and βIV spectrin through ankyrin G (Pan et al., 2006; Dzhashiashvili et al., 2007). In the peripheral nervous system (PNS), clustering of Na+ channels along the axon is controlled by the overlying Schwann cells (Eshed-Eisenbach and Peles, 2013). During development, Na+ channels initially cluster at heminodes that are formed at the end of each myelin segment (Vabnick et al., 1996; Ching et al., 1999; Schafer et al., 2006). These initial heminodal clusters will eventually give rise to mature nodes (Dugandzija-Novaković et al., 1995; Vabnick et al., 1996).
PNS node assembly is governed by two independent, yet cooperative mechanisms: clustering of Na+ channels at heminodes and mature nodes and restriction of their distribution between two paranodal junctions (Feinberg et al., 2010). These two mechanisms require different adhesion molecules and cytoskeletal proteins that are present at the nodes and adjacent paranodes. At mature nodes, the paranodal junctions serve as a membrane barrier that limits the lateral diffusion of nodal Na+ channels (Rosenbluth, 1976; Rasband et al., 2003; Rios et al., 2003; Zhang et al., 2013). The paranodal junction barrier mechanism also plays a major role during the formation of nodes in the CNS (Sherman et al., 2005; Susuki et al., 2013).
At the nodes, axon-glia interaction is mediated by binding of NF186 to gliomedin, a secreted ECM protein that is present with glial NrCAM at the Schwann cell microvilli that contact the nodal axolemma (Eshed et al., 2005; Eshed et al., 2007; Feinberg et al., 2010). Binding of gliomedin to NF186 induces the formation of node-like clusters containing Na+ channels, ankyrin G, and βIV spectrin (Eshed et al., 2005) and targets NF186 to heminodes (Zhang et al., 2012). Genetic deletion of gliomedin, glial NrCAM, or axonal NF186 completely abolishes the clustering of Na+ channels at heminodes (Feinberg et al., 2010), but not the formation of mature nodes due to the compensating restriction mechanism provided by the paranodal junction (Sherman et al., 2005; Feinberg et al., 2010). In the present study, we examine the role that nodal axon-glia interactions play after the nodes have already formed. We show that, in addition to the functions that gliomedin and NrCAM play during development, they also contribute to the long-term maintenance of Na+ channels at nodes of Ranvier.
Materials and Methods
Generation of mutant mice.
Generation of gliomedin mutant mice was described previously (Feinberg et al., 2010). Heterozygous gldn−/+ and nrcam−/+ (Custer et al., 2003) mice were crossed to generate double mutant mice. At least three mice of either sex of each genotype were used for immunofluorescence and EM analysis per age group. All experiments were performed in compliance with the relevant laws and institutional guidelines and were approved by the Weizmann Institute's Animal Care and Use Committee.
RNA preparation and RT-PCR.
Sciatic nerves were dissected from wild-type and mutant mice and immediately placed in QIAzol Lysis Reagent (QIAGEN). Total RNA was isolated using the RNeasy Mini Kit (Fatty Tissue Protocol; QIAGEN). cDNA was synthesized using SuperScript II Reverse Transcriptase (Invitrogen). Amplification of the desired fragments by PCR was achieved using the following primers: actin, 5′-GAGCACCCTGTGCTGCTCACCGGG-3′ and 5′-GTGGTGGTGAAGCCCC CTGTAGCCACGCT-3′; gliomedin, 5′-TACGCTTCAAGTGTGGATGGC-3′ and 5′-CAGGAT AGAGCATCAGATGGC-3′; and NrCAM, 5′-AGTACCCCTATTTGCTGAAGGC-3′ and 5′-CG CTGATCATCTGGCACAGG-3′.
Behavioral tests.
To evaluate motor impairments in wild-type and mutant mice, postnatal day 60 (P60) animals were subjected to a set of behavioral tests. Motor reflexes and gross body coordination were assessed using the tail suspension test. Mice were tested by evaluating their ability to walk across horizontal bars and by subjecting them to a bar test, during which they were left to hang from a stationary rod and the time elapsed until they fell was measured. Eight animals of each genotype were used throughout the tests.
Electrophysiology.
Sciatic nerve conductivity measurements were performed essentially as described previously (Poliak et al., 2003). Briefly, sciatic nerves were dissected and placed in a temperature-controlled recording chamber set to 37°C. The ends of the nerves were drawn into suction electrodes for stimulation and recording of compound action potentials. Signals were amplified, digitized, recorded, and analyzed on a laboratory computer using a pClamp10 program (Molecular Devices).
Tissue handling and immunofluorescence labeling.
Teased sciatic nerves and frozen sections were prepared and immunolabeled as described previously (Eshed et al., 2005; Feinberg et al., 2010). Briefly, sciatic nerves were dissected and immersed in 4% paraformaldehyde, pH 7.4, in PBS for 10–30 min at 4°C (depending on the age of the animal). Nerves intended for sectioning were cryoprotected in 20% sucrose (in PBS) overnight at 4°C, subsequently embedded in optimal cutting temperature medium (Tissue-Tek), and frozen on dry ice. Nerves intended for teasing were desheathed and teased using fine forceps on SuperFrost Plus slides (Menzel-Gläser; Thermo Scientific), air dried overnight, and then kept frozen at −20°C till use. When staining for protein zero (P0) or neurofilament H, samples were postfixed for 2–5 min using cold methanol (−20°C) and washed with PBS. When staining for neurofascin155, nerves were postfixed for 1 min in Bouin's fixative (Sigma-Aldrich) instead. Samples were incubated for 45 min in blocking solution (5% fish skin gelatin or normal goat serum and 0.5% Triton X-100 in PBS) at ambient temperature. If no postfixation was performed, blocking was elongated to 1 h. When staining for protein 4.1B, samples were left in blocking solution for 2 h. Samples were routinely incubated overnight at 4°C with the relevant mixture of primary antibodies diluted in blocking solution (5% fish skin gelatin or normal goat serum and 0.1% Triton X-100, in PBS). Nerve samples were then washed in PBS and incubated with secondary antibodies for 45 min at ambient temperature (same blocking solution as for the primary antibodies). Samples were washed again in PBS and then mounted with elvanol. Rabbit polyclonal antibodies against gliomedin (Eshed et al., 2007), Caspr (Peles et al., 1997), protein 4.1B (Gollan et al., 2002), neurofascin-155 (Poliak et al., 2001); rat antibody against Caspr (Feinberg et al., 2010); and mouse monoclonal antibodies against gliomedin (Eshed et al., 2005) and Caspr (Poliak et al., 1999) were described previously. Mouse monoclonal antibody against pan-sodium channels (S8809, clone K58/35) was purchased from Sigma Aldrich. Mouse monoclonal antibody against ankyrin G (clones N106/36 and 106/65) was purchased from NeuroMAb. Rabbit antibody against NrCAM (ab24344) was purchased from Abcam. A rabbit antibody against Phospho-Ezrin (Thr567)/Radixin (Thr564)/Moesin (Thr558; pERM, catalog #3141) was purchased from Cell Signaling Technology. A rat antineurofilament H monoclonal antibody (MAB5448) was purchased from Millipore. A chicken polyclonal antibody against protein zero (PZO) was purchased from Aves Labs. A mouse monoclonal antibody against neurofascin (IgM, A4/3.4) and a rabbit polyclonal antibody against βIV spectrin (C9831) were a gift from Dr. M. Rasband (Baylor College of Medicine, Houston, TX). A rabbit antibody against neurofascin-186 (MNF2) was kindly provided by Dr. P. Brophy (University of Edinburgh, Edinburgh, UK). Fluorescence images were obtained using an Axioskop2 microscope equipped with an ApoTom imaging system (Carl Zeiss) fitted with a Hamamatsu ORCA-ER CCD camera. Images were acquired and processed using the Zen2012 software (Carl Zeiss).
Electron microscopy.
Sciatic nerves of wild-type and mutant mice were exposed and fixed by continuous dripping of fresh fixative for 40 min (fixative containing 4% paraformaldehyde, 2.5% glutaraldehyde, and 0.1 m sodium cacodylate, pH 7.4, in PBS). Sciatic nerves were then carefully dissected, placed in fixative, and left to rotate overnight at ambient temperature while protected from light. Samples were then transferred to 4°C until processing. Processing was performed as described previously (Novak et al., 2011). Sections were subsequently examined using a Philips CM-12 transmission electron microscope.
Quantitation and statistical analysis.
All quantitation was performed on 12-μm-thick cryosections of sciatic nerves. Samples from at least three animals were used per genotype per age and counts were made from 10 fields of view per sample. Statistical significance was determined using Student's two-tailed t test.
Results
Mice lacking both gliomedin and NrCAM exhibit neurological abnormalities and slower nerve conduction
Because both gliomedin and NrCAM interact directly with NF186 (Eshed et al., 2005) and could thus potentially provide redundant functions in mature nodes, we generated mutant mice lacking both genes (Fig. 1A,B). Homozygous gldn−/−/nrcam−/− mice were born at the expected Mendelian ratios. In contrast to single mutant mice, which are by and large undistinguishable from wild-type mice, gldn−/−/nrcam−/− mice demonstrated major neurological impairments. Double mutant mice were smaller than their control littermates and displayed motor abnormalities that varied in severity between individuals. It ranged from strong ataxia, uncoordinated movements, and premature death to weakness of hind limbs and hypomotility followed by recovery at 3 weeks of age. However, despite their apparent amelioration, gldn−/−/nrcam−/− mice were not fertile and the majority died by P60. Double homozygous mutant mice displayed enhanced limb-clasping reflexes when suspended by the tail, unlike wild-type mice or the single mutants, which extended their limbs (Fig. 1C). When left to hang from a stationary rod, double mutant mice fell within seconds, whereas their control littermates held for more than a minute (Fig. 1D). Double gldn−/−/nrcam−/− mice had difficulty crossing horizontal bars, stumbled (Fig. 1E), and exhibited a wider gait, measured as a larger distance between ipsilateral limbs when walking (data not shown). The observed motor impairments correlated with reduced nerve conduction velocity along their sciatic nerves (Fig. 1F). Nerves of gldn−/−/nrcam−/− mice had a significantly slower propagation of action potential (13.4 ± 2 m/s) compared with wild-type (19.8 ± 3 m/s) and single mutant mice (gldn−/− 20.7 ± 4 m/s; nrcam−/− 19.6 ± 2 m/s). The refractory period of these nerves was also affected, being 30% longer than that measured in the controls (data not shown). Analysis of sciatic nerve cross-sections by electron microscopy revealed the presence of normal myelin morphology in all four genotypes (Fig. 1G), suggesting that the motor dysfunction and reduced nerve conduction may reflect nodal abnormalities.

Absence of gliomedin and NrCAM results in motor abnormalities. A, RT-PCR analysis of sciatic nerve mRNA using primer pairs specific to gliomedin and NrCAM revealed the absence of both transcripts in double homozygous mutant mice (gl/nr−/−). Primers for actin were used as controls. Size markers are given in base pairs. B, Gliomedin and NrCAM are undetectable in sciatic nerves of the double mutant mice. Teased sciatic nerves isolated from P60 mice of the indicated genotypes were immunolabeled using antibodies to gliomedin (Gldn), NrCAM, and P0 protein (P0). The location of the node in the double mutant is marked with an asterisk. Scale bar, 10 μm. C, Double mutant mice exhibit abnormal clasping of their hind limbs when suspended by their tails. Note the normal hind legs spreading of the single mutant animals. D, Adult double mutant mice (nrcam−/−/gldn−/−) exhibit a shorter latency to fall when left to hang from a horizontal rod by their forelimbs. Note that >80% of the double mutants fell after <5 s compared with wild-type mice, which stayed on the bar for more than a minute (n = 8). E, The double mutants stumble when placed on horizontal bars, whereas their single mutant littermates were undistinguishable from wild-type mice (nrcam−/−, bottom, and data not shown). F, Compound action potentials recorded from P15 mice show a reduction in nerve conduction velocity in the double but not single mutant mice. Error bars indicate SEM of n > 7 mice per genotype; p < 0.001. G, Electron microscopy images of sciatic nerve cross sections obtained from P120 wild-type (WT) mice, NrCAM-null (nrcam−/−), gliomedin-null (gldn−/−), or double mutant (gldn−/−/nrcam−/−) mice showing normal myelin. Scale bar, 10 μm.
Absence of the glial clustering signal leads to a gradual loss of nodal Na+ channels
Immunolabeling of sciatic nerves isolated from adult (P120) gldn−/−/nrcam−/− mice using antibodies to Na+ channels and Caspr revealed that some nodes (defined as the gap between Caspr-labeled paranodes) are devoid of sodium channels (Fig. 2A). Nodes lacking Na+ channel clusters were distributed sporadically and were often detected along axons that contain normal nodes as well (data not shown). Quantitation of the number of nodes lacking Na+ channels revealed that, at P15, ∼20% of the nodes lacked detectable Na+ channel clusters (Fig. 2B, top). However, at P60, nearly all nodes in the double mutant contained Na+ channels (Fig. 2B, middle). Accordingly, at P60-P75, no significant difference in nerve conduction velocity was detected in the single (gldn−/− 35.6 ± 3.6 m/s; nrcam−/− 37.7 ± 4.3 m/s), double heterozygous (gldn−/+/nrcam−/+ 34 ± 3.1 m/s), or double homozygous mutant mice (gldn−/−/nrcam−/− 33.3 ± 5.7 m/s). At P120, high-density accumulation of Na+ channels was detected at 69% of nodes in the double mutant, compared with almost all of the nodal gaps in the wild-type, gldn−/−, and nrcam−/− nerves (Fig. 2B, bottom). The latter reduction in the number of nodes containing Na+ channels at P120 suggests that, once accumulated, gliomedin and NrCAM play a role in maintaining them at the nodes. Furthermore, the finding that Na+ channels are still present in older single gldn−/− or nrcam−/− mice indicates that gliomedin and NrCAM have a redundant maintenance function in mature nodes.
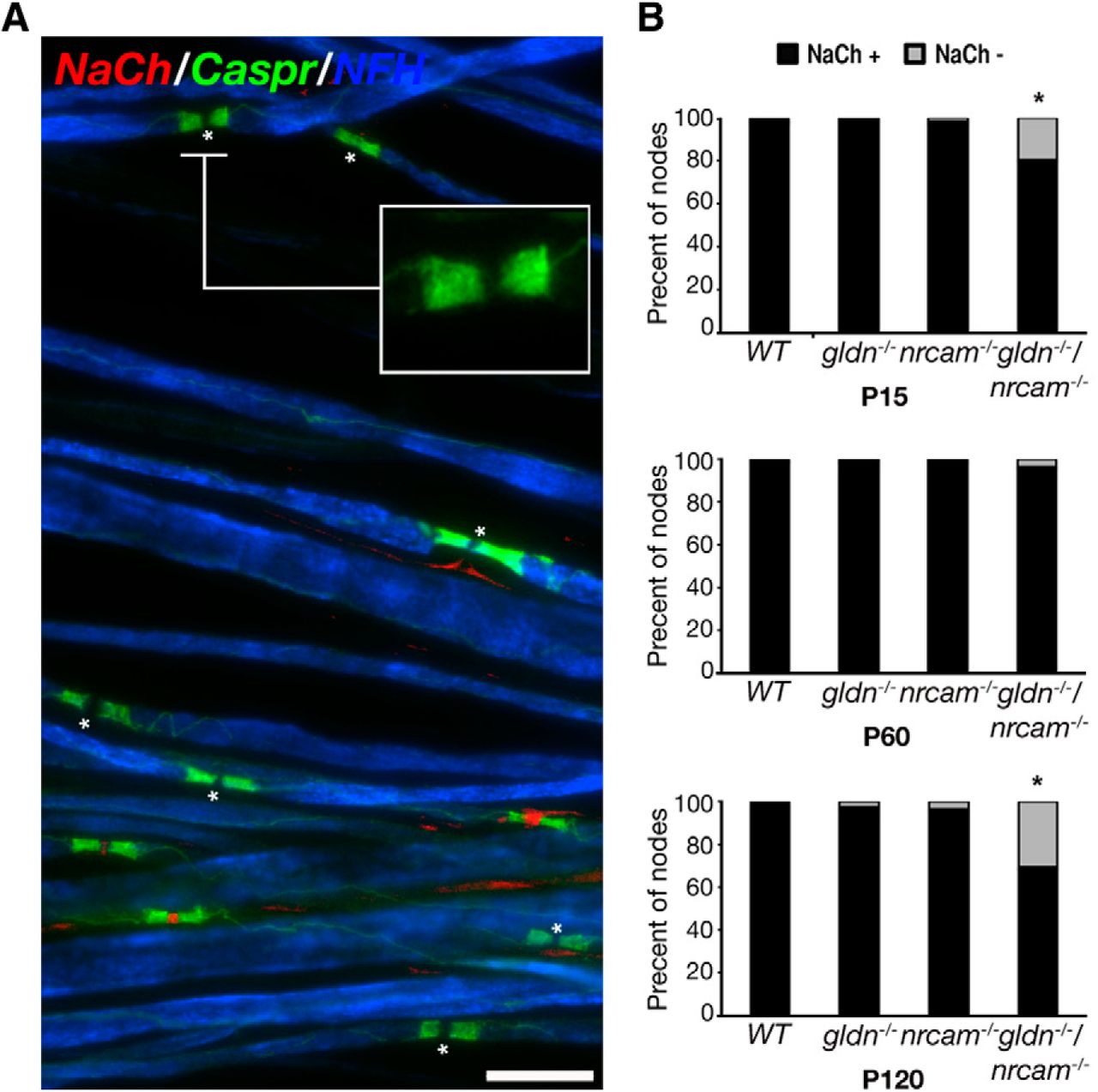
Disappearance of nodal Na+ channels in double gldn−/−/nrcam−/− mutant sciatic nerves. A, Immunolabeling of teased sciatic nerve fibers isolated from adult (P120) double mutant, showing nodes of Ranvier that lack Na+ channels (asterisks). Immunolabeling using antibodies to Caspr and neurofilament (NF-H) was performed to label the paranodal junction and the axon, respectively. B, Quantitative analysis of sciatic nerves isolated at P15, P60, and P120 from wild-type (WT), gliomedin-null (gldn−/−), NrCAM-null (nrcam−/−), and double mutant (gldn−/−/nrcam−/−) mice. Percent of nodes lacking Na+ channels is shown in gray; nodes containing Na+ channels are in black. More than 300 nodes from 3–5 animals per genotype were analyzed, *p < 0.0001. Scale bar, 10 μm.
In contrast to wild-type or the single mutants, Na+ channels in double gldn−/−/nrcam−/− mice were often concentrated near the paranodes, leaving a bare gap in the middle of the node; therefore, we termed these “binary nodes” (Fig. 3A–D). We then determined the incidence of binary nodes in P15, P60, and P120 sciatic nerves of all genotypes (Fig. 3E). In the wild-type nerves, <1% of all nodes appeared as binary at all ages. In the single gldn−/− and nrcam−/− mutants, binary nodes could be observed only in the adult nerves, where they accounted for <3% of all Na+ channel-positive nodes. In the double gldn−/−/nrcam−/− mutant at P15, as in the control samples, only <1% appeared binary. However, at P60, nearly 15% of Na+-channel-positive nodes were binary, a value that almost doubled by P120 (27%). In addition to Na+ channels, NF186, βIV spectrin, and ankyrin G were also affected and appeared as binary in adult (P120) mice (Fig. 4). In contrast, pERM immunoreactivity was always detected as a single stripe, an observation that is consistent with a previous study showing that it accumulates in Schwann cell distal processes once they overlay the axon (Gatto et al., 2003). Therefore, the absence of gliomedin and NrCAM affects the distribution of axonal components of the node of Ranvier, including channels, adhesion molecules, and cytoskeletal proteins.

Double mutant mice exhibit binary Na+ channel clusters and widening of the nodal gap. A, Immunofluorescence labeling of teased sciatic nerve isolated from a P120 double mutant mouse using antibodies to Na+ channels (NaCh) and Caspr. At this age, Na+ channels were either present (arrowhead), absent (arrows), or appeared as binary clusters (two small arrowheads) at the nodal gap. B–D, Additional examples of binary Na+ channel clusters in the double mutant (Na+ channels, green; Caspr, red). A line scan representing the intensity of the Na+ channel fluorescence signal (arbitrary units) along the nodal area is shown on the right of each panel. E, Increased appearance of binary nodal Na+ channels with age. Quantitation of the number of nodes containing binary Na+ channel clusters in sciatic nerves of wild-type (WT), single (gldn−/−, or nrcam−/−), or double (gl/nr−/−) mutant mice at P15 (2w), P60 (2m), and P120 (4m). Data are presented as a percentage of all Na+-channel-positive nodes. Error bars indicate SD of n = 3–5 animals per age per genotype (300 nodes per group), *p < 0.001. F, Double mutant nodes of Ranvier are longer. P60 or P120 sciatic nerves, isolated from the indicated genotypes, were labeled using antibodies to Caspr to demarcate the nodal gap. An asterisk marks a longer nodal gap in the double mutant. The percent of nodes that are longer than 1.3 μm and 1.6 μm in P60 and P120 mice, respectively, is shown. Error bars indicate SD of n = 3–5 animals per genotype per age (counted at least 300 sites per group), *p < 0.005. Scale bars, 5 μm.

Molecular composition of the nodes in the absence of gliomedin and NrCAM. Immunolabeling of teased sciatic nerve fibers isolated from adult wild-type (WT) and double mutant mice (gl−/−/nr−/−) using antibodies to neurofascin 186 (NF186) and Na+ channels (NaCh; A), βIV-spectrin and ankyrin G (AnkG; B), or to phospho-ERM (pERM) and Na+ channels (C and insets). Merged images are found on the right of each panel. Note the segregation of NF186, ankyrin G, and βIV-spectrin, but not of pERM, with nodal Na+ channels. Scale bar, 5 μm.
The observation that axonodal components disintegrate and form two separate bands prompted us to measure the length of the nodal gap using longitudinal sections of adult sciatic nerves from all genotypes (Fig. 3F). For each age, we set a threshold based on the length of the nodal gap in wild-type animals (i.e., 1.3 μm at P60 and 1.6 μm at P120). As depicted in Figure 3F, we found that ∼25% of all double mutant nodes are longer than the set threshold (24.05 ± 10.4% and 24.28 ± 8.85% of nodes at P60 and P120, respectively). This was in contrast to P15 animals, in which there was no significant difference in nodal length between the single and the double mutant mice (data not shown). Together, our results demonstrate that the absence of both gliomedin and NrCAM results in nodal disintegration, loss of Na+ channels, and dysregulation of the nodal gap length. They also indicate that the presence of only one of these molecules is sufficient to maintain axonal proteins at the nodes of Ranvier.
Absence of gliomedin and NrCAM results in abnormal node-paranode boundary
To extend these findings, we examined the morphology of the nodal region in the gldn−/−/nrcam−/− mutant by electron microscopy and compared it with the wild-type and single mutant animals. Longitudinal sections of sciatic nerves isolated from P120 wild-type mice revealed a clear border between the paranodal junction and the nodal gap, which is filled with microvilli that emanate from the outer aspect of the Schwann cell (Fig. 5A). The single gldn−/− and nrcam−/− mutants exhibit normal paranodal junctions that are sharply demarcated from the nodal gap, but Schwann cell microvilli are reduced in number (Fig. 5B,C). In agreement with our previous observations (Feinberg et al., 2010), the nodal gap is of normal length and, when present, Schwann cell microvilli are found outside the nodal axolemma and are disoriented rather than being perpendicular to the axis of the axon. At the double gldn−/−/nrcam−/− mutant nodes, Schwann cell microvilli are sparse or absent and both Schwann cell terminal loops and the outermost layer of the Schwann cell tend to overgrow along the nodal axolemma, sometimes largely occluding the nodal gap (Fig. 5D). Remarkably, Schwann cell processes that run parallel to the nodal gap form focal paranodal junction-type contacts with the axolemma (Fig. 5D, arrowhead, Fig. 6D–F). The appearance of these junctional contacts within the nodal gap correlated well with the presence of Caspr, as well as NF155 and protein 4.1B, at nodes containing binary clusters of Na+ channels, as detected by immunofluorescence labeling (Fig. 7).

Absence of gliomedin and NrCAM results in abnormal nodal morphology. Electron micrographs of sciatic nerves showing the node-paranode area in WT (A), nrcam−/− (B), gldn−/− (C), and gldn−/−/nrcam−/− double mutant mice (D). The location of the nodal gap is marked by arrows. In WT mice (A), the node is surrounded by abundant microvilli (MV). In the single mutant (B–C), and more profoundly in the double mutant (D), nonmicrovillar Schwann cell processes contact the nodal axolemma (asterisks). In the double mutant,these processes occasionally form a paranodal-junction-like kissing point with the axon (arrowhead in D). Scale bars, 1 μm.

Double gldn−/−/nrcam−/− mutant mice exhibit abnormal nodal boundaries and show junctional paranodal contacts within the nodal gap. Electron micrographs of the nodal region in sciatic nerves isolated from the double mutant. A–C, Three examples of nodes in which microvilli are absent and the outermost layer of the Schwann cell (asterisks) extends into the nodal gap and under the paranodes, intercalating itself between the axolemma and several terminal loops. D–F, The double mutant nerves exhibit junctional contact sites within the nodes. D, The node is elongated (∼1.5 μm; arrows), the paranodal terminal loops are present, and nodal microvilli are absent. The outer layer of the myelin segment at left extends a process that approaches the axolemma and then divides into two branches. One extends under terminal loops of the left paranode (asterisks) and the other extends over the node toward the adjacent myelin segment at right. The latter branch is separated from the axolemma by a wide gap along most of the node, but forms a paranode-type junction at which the apposed membranes approximate each other closely (arrowheads). E–F, High magnification of the boxed area in D showing a paranode-type junction at which the apposed membranes are separated by only 2–4 nm (arrowheads). The junctional gap at these sites is irregularly dense, but discrete transverse bands cannot be resolved because of the angle of the section. G, Nodal gap overhung by myelin lamellae and terminal loops primarily from the Schwann cell at left. The nodal axolemma appears dense because of a cytoskeletal “undercoating.” The loops overhanging the node do not indent the axolemma or form paranodal-type junctions with it, but markedly reduce the apparent length of the nodal gap. Schwann cell microvilli are virtually absent. H, Adjacent myelin segments form terminal loops (asterisks) that terminate against the axon very close to one another, leaving a very short (0.1 μm) nodal gap (arrowhead). Scale bars: A–D, 1 μm; E, F, 0.2 μm; G, H, 1 μm.

Inclusion of paranodal junction components within nodes of Ranvier of double mutant mice. A–C, Caspr immunoreactivity is detected within the nodes. Teased sciatic nerves isolated from P120 double mutants were labeled using antibodies to Na+ channels (NaCh) and Caspr (A), Caspr (B), or Caspr, Na+ channels, and βIV spectrin (C). Higher magnification of the boxed area in A is shown on the right. Asterisks mark the location of Caspr within the nodes, which is flanked by binary clusters of Na+ channels and βIV spectrin. D, E, Nodal Caspr colocalized with glial neurofascin 155 (D) and the adaptor protein 4.1B (E). Note that, similar to Caspr, NF155 and 4.1B are located at the nodal areas that lack Na+ channel clusters (asterisks). Scale bars: A–C, 5 μm; D, E, 10 μm.
In contrast to the single mutants, in double gldn−/−/nrcam−/− mice, Schwann cell processes sometime intercalate themselves between the paranodal terminal loops and the axolemma, undercutting them and hence lengthening the nodal domain (Fig. 6A–C). Intrusion of Schwann cell processes into the paranodes mainly affected the area proximal to the node, leaving the distal part of the paranodal junction intact (and thus positive for Caspr; Figs. 2, 3, 7). We also noted cases in which some of the paranodal loops invaded the nodal gap but did not form paranodal-type junctions with the nodal axolemma (Fig. 6G). Lastly, the double mutants also exhibit nodal gaps that lack Schwann cell microvilli and in which the terminal loops from the flanking paranodes terminate near each other against the axon, leaving only a minuscule nodal interval between them (Fig. 6H). Therefore, a lack of axo-glial contact at the node in double gldn−/−/nrcam−/− mice resulted in a variety of nodal-paranodal structural abnormalities.
Disintegration of the nodal complex reflects its sequence of assembly during development
To determine the sequence of events leading to the disintegration of the nodes of Ranvier in the absence of gliomedin and NrCAM, we examined whether the disappearance of Na+ channels from the nodes could be correlated with the absence of NF186, ankyrin G, or spectrin βIV from these sites. Notably, NF186 is of particular interest because it serves as an axonal receptor for gliomedin and NrCAM and is necessary for the maintenance of sodium channels at the axon initial segment (AIS; Zonta et al., 2011). Double immunolabeling of sciatic nerves isolated from P120 gldn−/−/nrcam−/− mice using antibodies to Na+ channels and NF186 (Fig. 8A), AnkG (Fig. 8B), or βIV spectrin (Fig. 8C) revealed differential disappearance of the different proteins from the nodes. For example, we detected nodes that contained Na+ channels, but lacked NF186 (Fig. 8A); the opposite situation was not observed at any point. AnkG was present in virtually all Na+ channel-containing nodes (Fig. 8B), whereas βIV spectrin was found at nodes that lacked these channels (Fig. 8C). In addition, the disappearance of βIV spectrin from nodes was invariably associated with the absence of NF186, AnkG, and Na+ channels (data not shown). As depicted in Figure 8D, at P60, nearly all nodes contained Na+ channels, yet 22% of the sites lacked NF186. At P120, Na+ channels were present in 69% of the nodes, whereas only 56% of the nodes stained positive for NF186. Furthermore, comparing the mutual presence of Na+ channels and NF186 revealed that a fifth of the nodes that contained Na+ channels lacked NF186 (18.8 ± 6.7% and 19.6 ± 5.4% at P60 and P120, respectively). Therefore, the loss of Na+ channels from nodes of Ranvier in gldn−/−/nrcam−/− nerves is preceded by a steady and gradual disappearance of NF186 from the axolemma. Similar to Na+ channels, ankyrin G and βIV spectrin were present in most of the nodes at P60, but at P120, 26% of the nodal gaps lacked ankyrin G and 10% were missing βIV spectrin (Fig. 8D). Together, our results suggest a progressive disassembly of the nodal complex, starting with the disappearance of NF186, followed by the loss of Na+ channels and ankyrin G and then βIV spectrin (Fig. 8E). Interestingly, this sequence of events reflects the assembly of nodes during development.

Sequential disappearance of nodal components reflects their order of assembly. A–C, Sections of sciatic nerve isolated from a P120 double mutant mouse were immunolabeled using antibodies to Na+ channels (NaCh) and NF186 (A), ankyrin G (B), or βIV spectrin (C). Note the presence of nodes that lack NF186 but still contain Na+ channels (A), as well as nodes that contain βIV spectrin but lack Na+ channels. Also note that ankyrin G and Na+ channels disappear from the nodes together. Scale bar, 5 μm. D, Percentage of nodes lacking NF186, Na+ channels, ankyrin G, and βIV spectrin in sciatic nerves isolated from gldn−/−/nrcam−/− mutant mice; n = 3 animals per genotype per age (300 sites counted per group), p < 0.005. E, Schematic summary of node disassembly in the absence of gliomedin and NrCAM (a–d). Loss of NF186 precedes that of Na+ channels and AnkG, which occurs before the loss of βIV spectrin from the nodes.
Discussion
Nodes in the PNS are assembled by two cooperating mechanisms: clustering of nodal components at heminodes and their restriction to the nodal gap by the adjacent paranodal junction (Feinberg et al., 2010). Initial clustering of Na+ channels occurs at heminodes and is formed at the edge of myelin segments. This process is mediated by binding of Schwann cell-derived gliomedin and NrCAM to their axonal receptor NF186 (Eshed et al., 2005; Feinberg et al., 2010). Gliomedin is localized to the Schwann cell microvilli by glial NrCAM (Feinberg et al., 2010) and by its association with the surrounding nodal ECM (Eshed et al., 2007). The concentration of gliomedin by NrCAM and the nodal ECM forms high-avidity complexes that trap NF186 and cluster it on the underlying axolemma (Zhang et al., 2012). In the absence of a functioning heminodal clustering mechanism (e.g., in mice lacking gliomedin or NrCAM; Feinberg et al., 2010), node assembly is driven solely by the flanking paranodal junctions, which serve as diffusion barriers that limit the lateral movement of membrane proteins (Rosenbluth, 2009). In agreement, we found that nodes did form in the absence of both gliomedin and NrCAM, further supporting a primary role for the paranodal-junction-dependent mechanism in the assembly of PNS nodes. Nevertheless, the delay in the accumulation of Na+ channels in mature nodes in the double gldn−/−/nrcam−/− mutant, together with the normal appearance of nodes in paranodal mutant mice (Bhat et al., 2001; Boyle et al., 2001; Gollan et al., 2003), indicate that, in the PNS, the paranodal restriction mechanism is not as efficient as the heminodal clustering of sodium channels.
We show that the absence of both gliomedin and NrCAM led to gradual disintegration of the nodes, as evident by the loss of Na+ channels and other nodal components. These results show that Schwann cell-axon contact mediated by gliomedin, NrCAM, and NF186 not only promotes the clustering of Na+ channels, but also prevents the nodes from dissociating. They further indicate that although PNS nodes could be formed by the paranodal-junction-dependent barrier, this mechanism is not always sufficient to maintain Na+ channel clusters at the axolemma in mature nodes. It was shown recently that although the initial accumulation of NF186 at heminodes requires its extracellular domain, its cytoplasmic ankyrin-G-binding domain mediates its transport and stabilization in mature nodes (Zhang et al., 2012). Our results indicate that, in addition to ankyrin G, continued interaction of NF186 with its glial ligands is required for the stabilization and maintenance of nodes of Ranvier. Persistent interaction of NF186 with both cytoskeletal components and extracellular proteins localized to the nodal gap may contribute to the extremely long half-life of this adhesion molecule in myelinated axons (Zonta et al., 2011; Zhang et al., 2012). This is further supported by the observation that the turnover of NF186 and Na+ channels is much slower at the nodes than at the AIS (Zonta et al., 2011). Such a mechanism is reminiscent of CNS nodes, in which binding of multiple ECM components to NF186 stabilizes the nodal complexes (Susuki et al., 2013).
Genetic ablation of gliomedin or NrCAM, which abolished heminodal clustering (Feinberg et al., 2010), did not affect the maintenance of Na+ channels at mature nodes. Therefore, whereas both gliomedin and NrCAM are required for the clustering of Na+ channels during node formation, either one of these molecules is sufficient for nodal maintenance. The compensatory role of gliomedin and NrCAM in nodal maintenance is likely due to their ability to interact directly with NF186 (Volkmer et al., 1996; Lustig et al., 2001; Eshed et al., 2005; Labasque et al., 2011). NrCAM and gliomedin are present in mature nodes containing NF186 in gldn−/− or nrcam−/−, respectively (Feinberg et al., 2010). We propose that, in the double mutant, the absence of Schwann cell-axon contact that is normally mediated by gliomedin and NrCAM leads to destabilization of NF186 at the nodal axolemma and further loss of Na+ channels. This notion is well supported by several studies showing that axonal NF186 plays a key role in the assembly (Sherman et al., 2005; Zonta et al., 2008; Thaxton et al., 2011) and maintenance of nodes (Zhang et al., 2013), as well as in the maintenance of the AIS (Zonta et al., 2011).
The observation that Na+ channels were still present in approximately two-thirds of the nodes in adult mice lacking both gliomedin and NrCAM (this study), or after depletion of NF186 in the adult (accompanied paper by Desmazieres et al., 2014), may suggest the involvement of other glial-derived ligands in preserving the nodal complex. The nodal gap in the PNS contains several proteins that could cooperate with gliomedin and NrCAM in maintaining the composition of the nodal axolemma (Eshed-Eisenbach and Peles, 2013). Some candidates include the adhesion receptor dystroglycan (Saito et al., 2003; Occhi et al., 2005); the heparan sulfate proteoglycans (HSPGs) syndecan 3, syndecan 4 (Goutebroze et al., 2003; Melendez-Vasquez et al., 2005), and perlecan (Bangratz et al., 2012; the ECM components collagen XXVIII (Grimal et al., 2010) and collagen V (Melendez-Vasquez et al., 2005); and the gliomedin-related protein myocilin (Kwon et al., 2013). Gliomedin binds HSPGs (Eshed et al., 2007) and this interaction could further contribute to nodal stabilization in a manner similar to the role that HSPGs play in CNS nodes (Dours-Zimmermann et al., 2009; Bekku et al., 2010; Susuki et al., 2013). Two additional interesting candidates are dystroglycan, which is necessary for the clustering of Na+ channels at the nodes of Ranvier (Occhi et al., 2005), and myocilin, which was recently reported to bind both gliomedin and NrCAM and to cluster NF186 and Na+ channels when presented to cultured sensory neurons (Kwon et al., 2013).
The organization of the axolemma of myelinated nerves occurs sequentially (Poliak and Peles, 2003). In the PNS, clusters of NrCAM and NF186 are the first to be detected, followed by ankyrin G and Na+ channels (Lambert et al., 1997). Ankyrin G, which is recruited to NF186 (Dzhashiashvili et al., 2007), is further required for the arrival of βIV spectrin (Yang et al., 2007). Analysis of the nodal composition in gldn−/−/nrcam−/− mice at different ages revealed the existence of nodes containing Na+ channels, ankyrin G, and βIV spectrin, but not NF186. We also noted the presence of nodes missing Na+ channels and ankyrin G but still containing βIV spectrin. These results indicate that the disassembly of the nodal complex in the absence of gliomedin and NrCAM occurred in an orderly manner that reflects its sequence of assembly: first NF186, followed by ankyrin G and Na+ channels, and then βIV spectrin.
The absence of both gliomedin and NrCAM resulted in several nodal abnormalities: (1) the absence of Schwann cell microvilli or their presence only outside of the nodal axolemma, (2) Schwann cell processes that cover the nodal axolemma and run parallel to it, (3) elongated nodal intervals, (4) formation of paranodal junction-like “kissing” points between Schwann cell processes and the nodal axolemma, (5) narrowing of nodes by the adjacent paranodal loops, and (6) intercalation of Schwann cell processes between the nodal proximal paranodal loops and the axon. These results suggest that the axon-glia interaction mediated by gliomedin and NrCAM prevents the penetration of the paranodal loops into the nodal gap. This conclusion is further supported by previous observations showing that genetic ablation of their axonal receptor NF186 results in similar morphological abnormalities (Thaxton et al., 2011).
The paranodal junctions form a membrane barrier that separates the juxtaparanodal Kv1 channels from the nodal area (Bhat et al., 2001; Boyle et al., 2001; Gollan et al., 2003) and, in the CNS, they also maintain Na+ channels between two myelin internodes (Rios et al., 2003). However, in the PNS, the nodes are still preserved in the absence of these junctions, indicating that additional nodal components prevent them from disassembling. Our results suggest the involvement of gliomedin and NrCAM in the maintenance of PNS nodes. The absence of these molecules often leads to elongation of the nodal gap and the formation of binary clusters of Na+ channels. Because gliomedin and NrCAM are required for heminodal clustering during development, it is likely that these binary clusters form differently. One possibility is that they reflect the accumulation of nodal components, including βIV spectrin, near the paranodal cytoskeletal barrier that is formed by βII spectrin (Zhang et al., 2013). This may be similar to the submembranous cytoskeleton barrier generated by αII and βII spectrin at the AIS (Galiano et al., 2012). Interestingly, binary Na+ channel clusters were flanked by a distinct line containing Caspr and protein 4.1B, indicating that paranodal proteins are still excluded from areas containing βIV spectrin. The presence of Caspr and protein 4.1B within the nodal gap was accompanied by the formation of paranodal-junction-like contacts between the Schwann cell process and the axolemma. The focal accumulation of Caspr and protein 4.1B was associated with NF155, which serves as the glial ligand for the Caspr/contactin complex (Charles et al., 2002) and is present at both the paranodal loops (Tait et al., 2000) and the Schwann cell microvilli (Brown et al., 2001; Gunn-Moore et al., 2006).
In summary, our results suggest that the interaction of gliomedin, NrCAM, and NF186 is important for the maintenance of peripheral nodes of Ranvier. These findings are of particular interest given the nodal pathologies detected in antiganglioside-antibody-mediated neuropathies (recently termed nodo-paranodopathy; Uncini et al., 2013), as well as the presence of autoantibodies to these nodal CAMs in Guillain-Barre syndrome and chronic inflammatory demyelinating polyneuropathy (Prüss et al., 2011; Devaux, 2012; Ng et al., 2012; Susuki, 2013). The presence of such antibodies in animal models of these neuropathies leads to the disorganization of the nodes of Ranvier, formation of some binary nodes, and lengthening of the nodal gap, as we observe in our double mutant mice (Lonigro and Devaux, 2009). Finally, the length and complex geometry of myelinated nerves in the PNS requires efficient targeting and stabilization mechanisms to ensure their normal function. We suggest that continuous axon-glia interaction at the nodes constitute a part of such mechanisms.
Footnotes
This work was supported by the National Institutes of Health (Grant NS50220 to E.P. and Grant NS037475 to J.R.), the Israel Science Foundation, The Legacy Heritage Biomedical Fund, the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, and the National Multiple Sclerosis Society (Grant RG3618B9 to J.R.). E.P. is the Incumbent of the Hanna Hertz Professorial Chair for Multiple Sclerosis and Neuroscience. We thank Peter Shrager for discussion and valuable advice, Matthew Rasband and Peter Brophy for antibodies, and Raffi Saka for mice maintenance.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Elior Peles, Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot 76100, Israel. peles{at}weizmann.ac.il





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}