Article Text

Abstract
Mutations in the gene for fibrillin-1 (FBN1) have been shown to cause Marfan syndrome, an autosomal dominant disorder of connective tissue characterised by pleiotropic manifestations involving primarily the ocular, skeletal, and cardiovascular systems. Fibrillin-1 is a major component of the 10-12 nm microfibrils, which are thought to play a role in tropoelastin deposition and elastic fibre formation in addition to possessing an anchoring function in some tissues.
Fibrillin-1 mutations have also been found in patients who do not fulfil clinical criteria for the diagnosis of Marfan syndrome, but have related disorders of connective tissue, such as isolated ectopia lentis, familial aortic aneurysm, and Marfan-like skeletal abnormalities, so that Marfan syndrome may be regarded as one of a range of type 1 fibrillinopathies.
There appear to be no particular hot spots since mutations are found throughout the entire fibrillin-1 gene. However, a clustering of mutations associated with the most severe form of Marfan syndrome, neonatal Marfan syndrome, has been noted in a region encompassing exons 24 to 32. The gene for fibrillin-2 (FBN2) is highly homologous to FBN1, and mutations inFBN2 have been shown to cause a phenotypically related disorder termed congenital contractural arachnodactyly. Since mutations in the fibrillin genes are likely to affect the global function of the microfibrils, the term microfibrillopathy may be the most appropriate to designate the spectrum of disease associated with dysfunction of these molecules.
The understanding of the global and the molecular functions of the fibrillin containing microfibrils is still incomplete and, correspondingly, no comprehensive theory of the pathogenesis of Marfan syndrome has emerged to date. Many, but not all, fibrillin-1 gene mutations are expected to exert a dominant negative effect, whereby mutant fibrillin monomers impair the global function of the microfibrils. In this paper we review the molecular physiology and pathophysiology of Marfan syndrome and related microfibrillopathies.
- Marfan syndrome
- fibrillin
- microfibrillopathies
Statistics from Altmetric.com
Marfan syndrome
Marfan syndrome (MFS) is an autosomal dominant heritable disorder of connective tissue that involves primarily the skeletal, ocular, and cardiovascular systems. The leading cause of premature death in MFS patients is progressive dilatation of the aortic root and ascending aorta, causing aortic incompetence and dissection.1 2 MFS has an incidence of at least 1:10 000 and about 25-30% of cases represent new mutations.3
A wide variety of skeletal abnormalities occurs with MFS, including dolichostenomelia, arachnodactyly, scoliosis, chest wall deformity (pectus carinatum or excavatum), tall stature, ligamentous laxity, abnormal joint mobility, and protrusio acetabulae. Scoliosis is found in about 60% of adults of both sexes with MFS.4 Pectus excavatum of some degree of severity occurs in about two thirds of all MFS patients, and may have a greater tendency to recur after surgical repair than idiopathic pectus excavatum.5
Ectopia lentis occurs in up to about 80% of MFS patients and is almost always bilateral.6 The most common cardiovascular manifestations of MFS affect the atrioventricular valves and the aorta. Mitral valve disease may be the earliest of the cardiovascular manifestations of MFS. Progressive dilatation of the aortic root is responsible for most cases of aortic incompetence; usually, there is a gradual dilatation starting at the aortic root which may extend into the ascending aorta. This may then lead to the sudden onset of aortic dissection, which can cause acute aortic regurgitation or aortic rupture with sudden death.2 Generalised aortic root dilatation7 as well as a family history of severe cardiovascular disease in relatives with MFS8 are associated with a greater risk for aortic complications.
The average life expectancy has risen significantly since 1972,9 which is at least in part because of the benefits arising from cardiovascular surgery10 and medical therapy with beta blockers.11 12
The diagnosis of MFS can be made according to the criteria of the Gent nosology when major criteria are present in two organ systems and a third organ system is involved.13 Diagnostic dilemmas may arise because of the considerable inter- and intrafamilial variability of MFS. Also, many features of MFS, such as mitral valve prolapse or scoliosis, are common in the general population or may occur in other connective tissue disorders. Many manifestations are age dependent, and therefore the clinical criteria as defined in the Gent nosology cannot always be strictly applied to paediatric patients. This is particularly true in the case of children with sporadically occurring disease.14
The identification of the fibrillin-1 gene (FBN1)
Several strategies led to the identification of the fibrillin-1 gene (FBN1) in 1991. Fibrillin, a 350 kDa glycoprotein, was discovered in 1986 and characterised as the main component of the extracellular microfibrils.15 By means of immunohistochemical studies with monoclonal antibodies against fibrillin, it was shown in 1990 that most Marfan patients display abnormalities of the microfibrils in the extracellular matrix.16 A peptide from fibrillin was isolated from which a 20 amino acid sequence could be determined; this allowed appropriate combinations of PCR primers to be designed. One combination led to the amplification of a PCR product, which was used as a probe to isolate a 1.6 kb cDNA fragment belonging to the fibrillin-1 gene that was then assigned to chromosome 15q15-21 by in situ hybridisation.17 By this time, linkage analysis had mapped the MFS gene to the long arm of chromosome 15 in 1990,18 19 making the fibrillin-1 gene a promising candidate gene for MFS. Finally, Dietz et al 20 showed a de novo point mutation in the fibrillin-1 gene in two unrelated Marfan patients.
The structure of the fibrillin-1 gene
The fibrillin-1 gene (FBN1, previouslyFib15) spans about 200 kb genomic DNA21 with 65 exons and a transcript size of 10 kb. The mRNA possesses 9663 nucleotides with an open reading frame of 8613 nucleotides and 5′ and 3′ untranslated regions of 134 and 916 nt.22 Fibrillin-1, the protein product ofFBN1, is a cysteine rich monomeric glycoprotein with a molecular weight of about 350 kDa.23Profibrillin-1 contains 2871 amino acids and can be divided into a signal peptide for extracellular secretion and five structurally distinct domains22 (A-E). Domains B and D are composed of repeated motifs, which can be divided into three groups based on sequence homology (table 1). The first type of motif, termed epidermal growth factor-like motif (EGF), occurs 47 times in fibrillin-1; EGF motifs contain six highly conserved cysteine residues which form disulphide bridges with one another. Forty three of the 47 EGF repeats of fibrillin-1 also contain a calcium binding consensus sequence24 and are termed calcium binding EGF-like motifs (cbEGF). The second structural motif is analogous to a motif first described in the latent transforming growth factor-β1 binding protein (LTBP motif, also referred to as 8-cys or TGFβ1bp motif) and contains eight cysteine residues with an internal cluster of three consecutive cysteine residues. Seven LTBP modules occur in fibrillin domains B and D. The LTBP motifs display a globular structure and interrupt multiple tandem stretches of cbEGF modules that are thought to form rod-like structures.25 The fourth LTBP motif contains an RGD (arginine-glycine-aspartic acid) cell adhesion motif. RGD motifs interact with cell surface receptors of the integrin family to mediate cell adhesion. Integrins are transmembrane proteins which can interact with proteins of the extracellular matrix and with elements of the cytoskeleton, resulting in an anchoring of cells within the extracellular matrix (ECM).26 The RGD motif of fibrillin-1 was shown to promote cell adhesion via integrin ανβ3.27-29
Consensus sequences for motifs shared between the fibrillins and the LTBPs. Consensus sequences were determined from sequences taken from the references cited in fig 1. In the consensus sequences, X may be any residue and the numbers in parentheses indicate an average number of residues
The third structural motif may represent a fusion of portions of the EGF and LTBP motifs22 and has been termed the Fib motif. Like the LTBP motif, the Fib motif contains eight cysteine residues, two of which are clustered together in the same relative location as the three consecutive cysteine residues of the LTBP repeat. A nine cysteine derivative of the Fib motif is found in domain B.
Exon 1 contains the ATG start site for translation and codes for a signal peptide followed by a consensus processing sequence for propeptides RX(K/R)R↓; in vitro evidence suggests that fibrillin-1 undergoes intracellular N-terminal processing at this site.30 The cleavage site is followed by 12 mainly basic amino acids of unknown functional significance (domain A).
Domain B (exons 2-10) begins with a four cysteine motif with some similarities to the Fib motif, and also contains three non-calcium binding EGF-like motifs and the first two cbEGF motifs, a nine cysteine derivative of the Fib motif, and the first LTBP-like motif.
Domain C (exon 10) is characterised by an unusually high proline content (42%). It has been speculated that the high proline content may allow region C to bend in three dimensional space and act as a kind of molecular hinge.31
Domain D represents the largest single domain and comprises 2240 amino acids grouped in 49 cysteine rich repeats, comprising one non-calcium binding EGF-like motif and 41 cbEGF motifs, six LTBP-like motifs, and one Fib motif (exons 11-63).
The carboxy-terminal domain E (exons 64-65) represents a unique sequence which contains a pair of two consecutive cysteine residues within a sequence which is highly conserved between fibrillin-1 and fibrillin-2 (see below). This high level of conservation between the two fibrillins suggests that the region may possess a specific functional significance.32 In addition, the carboxy-terminus contains a stretch of basic amino acids harbouring a tetrabasic cleavage site (RKRR↓), which undergoes extracellular cleavage by an enzyme of the PACE (Paired basic Amino acid Cleaving Enzyme) convertase family.33-36
Fibrillin-1 contains 14 sites for N-linked glycosylation, the majority of which are located in domain D.22 Some, but not all, putativeN-glycosylation sites in normal profibrillin are used during its regular intracellular post-translational processing.37 Also, a specific Asn residue forming a part of this calcium binding consensus sequence is β hydroxylated.38
The fibrillin-2 gene (FBN2)
The fibrillin-2 gene (FBN2, previouslyFib5) on chromosome 5q23-q31 is closely related to fibrillin-1 (fig 1). The domain structure as well as the number and order of the sequence motifs are identical between the two proteins. Domains B and D of fibrillin-1 and -2 are 80% identical at the amino acid level. However, fibrillin-1 and -2 have important differences, which may reflect differing functional roles. Fibrillin-1 contains one RGD motif and fibrillin-2 contains two. Whereas domain C of fibrillin-1 is proline rich, domain C of fibrillin-2 contains a high content of glycine residues.32 Domains A and E are comparatively less related to the corresponding domains of fibrillin-1 (19% and 50% amino acid identity).

Domain structure of proteins of the fibrillin and latent transforming growth factor β1 binding protein (LTBP) families. Fibrillin-1,22 fibrillin-2,32LTBP-1S,194 LTBP-2,56 LTBP-3,59and LTBP-461 are shown. All proteins were sketched from the human sequence, except for LTBP-3, for which the human sequence has not yet been completely determined. In addition to the symbols shown in the figure, grey horizontal rectangles are used to denote sequences at least 40 amino acids long with an unusually high content of proline (P) or glycine (G); the approximate percentage of these residues is indicated underneath the corresponding rectangles. Horizontal lines denote unique sequences in the N- or C-terminal regions or elsewhere.
Fibrillin-1 and fibrillin-2 are differentially expressed, both in terms of developmental stages and tissue distribution. In most cases, developmental expression of the fibrillin genes exhibits a diphasic pattern, with the onset of FBN2 expression occurring earlier than FBN1 expression.FBN2 transcripts seem to accumulate before tissue differentiation and to decrease rapidly or disappear thereafter;FBN1 transcripts increase at a gradual rate thereafter. Fibrillin-2 is found preferentially in elastic tissues, such as the elastic cartilage, the tunica media layer of the aorta, and along the bronchial tree.39 The two fibrillins may therefore have differing functional roles; fibrillin-2 may possess a major functional role during early morphogenesis in directing elastic fibre assembly40; in contrast, the predominance of fibrillin-1 in stress and load bearing structures, like the aortic adventitia, the ciliary zonules, and the skin, suggests that fibrillin-1 may be mainly responsible for the structural function of the microfibrils.39
A second locus for MFS on chromosome 3?
Linkage to chromosome 3p24.2-p25 has been shown in a large family with a Marfan-like phenotype for whom linkage to the fibrillins had been previously excluded.41-43 While a cumulative lod score of >150 for linkage between fibrillin-1 and MFS makes genetic heterogeneity unlikely in classical MFS,44 it will not be surprising that mutations in other genes may cause non-classical Marfan-like phenotypes.
The latent transforming growth factor-β binding protein (LTBP) genes
The fibrillins are structurally related to the LTBP gene family. To date, four LTBP genes have been identified, all of which contain multiple tandem copies of the cbEGF motif as well as two different eight cysteine repeats found to date only in fibrillin and in the LTBPs. The first of these, the LTBP motif, contains eight cysteine residues with an internal cluster of three consecutive cysteines. The second motif, termed “Fib motif” because it was first described in fibrillin-1,22 contains eight cysteine residues with an internal cluster of two consecutive cysteines. The terminology surrounding these motifs can be confusing, especially since the LTBP motif and Fib motif have occasionally been lumped together as “8 cys repeats” or “TGF repeats”. Table 1 supplies a summary of the consensus sequences of the motifs found in the fibrillins and LTBPs. It is noteworthy that the order of the EGF motifs and the eight cysteine repeats in the LTBPs is similar to that in the fibrillins (fig 1).
LTBP-1 was initially characterised as a regulator of transforming growth factor-β (TGF-β) metabolism. TGF-β is a family of proteins that regulate growth, differentiation, adhesion, and migration of various cell types. Although TGF-β is growth inhibitory for most cell types, it can stimulate the production of extracellular matrix proteins, such as the collagens, fibronectin, elastin, and tenascin.
TGF-β is synthesised in a precursor form that undergoes self-association followed by endoproteolytic cleavage to form two disulphide bonded homodimers: mature TGF-β and the N-terminal propeptide of the TGF-β precursor. The N-terminal propeptide is termed latency-associated peptide (LAP), since it renders secreted TGF-β latent by remaining non-covalently associated with it to form the small latent TGF-β complex. The large latent complex additionally contains LTBP-1, a monomeric molecule that is covalently attached to the LAP by means of an exchange of cysteine disulphide bonds between the third LTBP (eight cysteine) repeat and the LAP during secretion.45 46
LTBP-1 undergoes rapid intracellular association with TGF-β, which augments the secretion of TGF-β from cells.47 Following secretion, LTBP-1 is able to store latent TGF-β in the extracellular matrix.48 49 LTBP-ECM interaction may be an important intermediate step in TGF-β activation.50 Release of TGF-β from the ECM as a consequence of proteolytic cleavage of LTBP-1 is one possible mechanism for the regulation of TGF-β action.51 52 LTBP-1 may also participate in the activation of latent TGF-β by concentrating it on the cell surface where activation occurs.53
In addition to its role as a regulator of TGF-β metabolism, there is evidence that LTBP-1 has a distinct role as an independent structural protein of the ECM. LTBP-1 and TGF-β1 co-localise not only with fibronectin and collagen type IV, but also with 10 nm diameter structures thought to represent fibrillin-associated microfibrils.54 The binding of LTBP-1 to the ECM is mediated by a covalent interaction localised to its amino terminal domain.45 An alternatively spliced form of LTBP-1, which is longer in its amino terminal domain (LTBP-1L), binds even more efficiently to the ECM.55
To date, little is known about the differential functional roles of the four LTBP isoforms, although LTBP-2, -3, and -4 share at least some of the functional properties of LTBP-1. LTBP-2 also forms a large, latent TGF-β complex and can associate with the ECM.56 Bovine LTBP-2 localises to elastin-associated microfibrils of the bovine nuchal ligament, and is bound covalently to the microfibrils by reducible disulphide linkages. Interestingly, the LTBP-2 was found not to bind to latent TGF-β, but appeared to be an independent structural component of the microfibrils.57 Like LTBP-1, LTBP-2 binds to the ECM via its amino-terminal domain and can be released from the ECM by proteolysis.58 Like LTBP-1 and -2, LTBP-3 can also bind to the TGF-β1 precursor,59 and this interaction also appears to be mediated by an LTBP motif.60 Finally, a fourth LTBP has been identified61 and appears also to undergo association with the extracellular matrix.62
Many of these microfibrillar elements localise to fibrillin-containing microfibrils. Therefore, they can be considered candidate genes for many of the disorders, named and unnamed, that clinically overlap with MFS. Preliminary studies based on such an approach are described later.
Elastic fibres, microfibrils, and fibrillin
The elastic fibre is a complex structure, which contains elastin, 10-12 nm microfibrils, lysyl oxidase, and perhaps proteoglycans. By electron microscopy, elastic fibres can be seen to be composed of two morphologically distinct components: an amorphous fraction lacking any regular structure and constituting about 90% of the mature fibre, which consists exclusively of elastin, and a microfibrillar component consisting of 10-12 nm microfibrils. Elastin-producing cells secrete tropoelastin, the 70 kDa monomeric precursor of elastin, into the extracellular space, where it is rapidly accreted to the surface of elastic fibres and becomes highly cross linked by the action of lysyl oxidase to form mature elastin. Elastin endows the elastic fibres with the property of elastic recoil, which is essential for the function of resilient tissues, such as the aorta, the lung, and the skin.63 The 10-12 nm microfibrils are located primarily around the periphery of the amorphous elastin component of the elastic fibres. The microfibrils display a “beads on a string” structure with a diameter of 8 to 12 nm and consist of several distinct proteins including fibrillin. Morphologically identical microfibrils have been found in association with elastin in a wide variety of tissues, including skin, lung, kidney, blood vessels, cartilage, and tendon. Microfibrils not associated with elastin may be observed in other tissues, such as the ciliary zonule.15
Whereas the role of elastin in mediating elastic recoil of tissues is well established, the functions of the microfibrils are not yet fully understood. The microfibrils do appear to subserve several global functions. First, they act as a scaffolding for tropoelastin deposition and elastic fibre formation.64 Second, microfibrils themselves are extensible,65 and they may contribute to the mechanical properties of mature elastic tissues by means of load redistribution between individual elastic fibres.66 Third, they provide structural anchorage in non-elastic tissues, such as the ciliary zonules.67 Fourth, microfibrils may serve to anchor endothelial and epithelial cells to elastic fibres of the ECM via cell binding domains.28 Fifth, a role for the microfibrils in the provision of a flexible mechanical anchor at epithelial-mesenchymal basement membrane interfaces, such as at the dermal epidermal junction, has been proposed.68 In addition, a role for fibrillin containing microfibrils in the support of platelet adhesion has been reported.69
In addition to fibrillin-1 and -2, a series of other proteins has been isolated from the microfibrils (table 2). Microfibril associated glycoprotein 1 (MAGP-1) is a 31 kDa glycoprotein consisting of two domains: an acidic N-terminal domain without cysteine residues and a basic C-terminal domain containing all 13 cysteine residues.70 MAGP-1 appears to be covalently bound to the microfibrils by intermolecular disulphide links71 72 and is also a substrate for transglutaminase,73 although it is not known which component of the microfibrils undergoes crosslinking with MAGP-1. Since the N-terminal region of MAGP-1 can undergo binding with both tropoelastin73 and type VI collagen,74 it is possible that MAGP-1 is responsible for the organisation of tropoelastin monomers in the ECM before cross linking,75 as well as playing a role in the interaction of fibrillin containing microfibrils and type VI collagen, which forms extensive networks of 3-5 nm microfibrils in virtually all soft connective tissues. MAGP-1 localises specifically to the “beads” of isolated microfibrils, and multiple MAGP-1 molecules may be present in a single bead.76
Protein components of fibrillin-containing associated microfibrils
The fact that dermal microfibrils contain gene products of bothFBN1 and MAGP-1is underlined by observations in early immunofluorescence studies of MFS patients. In these studies, abnormal (decreased) immunostaining results were noticed when antibodies to either fibrillin orMAGP-1 were used (Godfrey and Hollister, unpublished data). Thus MAGP-1 was as good a candidate as fibrillin in the quest for the aetiology of MFS. To date, however, there are no reports of mutations inMAGP-1.
MAGP-2 is a 25 kDa glycoprotein that is also disulphide-bonded to fibrillin containing microfibrils.77 There is a close similarity between MAGP-1 and MAGP-2 confined to a central region of 60 amino acids in the C-terminal domain including a precise alignment of seven cysteine residues.78 MAGP-2 does not appear to possess tropoelastin or type VI collagen binding properties, but instead possesses an RGD cell adhesion motif in its N terminal domain which can interact with a range of cell types via ανβ3 integrin.79 MAGP-2 has a more restricted tissue and developmental distribution than MAGP-1, which may reflect its presumed differing functional roles.80
The microfibril-associated protein 1 (MFAP-1) is a 58 kDa protein that is apparently post-translationally cleaved to a 32 kDa fragment that becomes an integral component of microfibrils.81 MFAP-1 does not appear to share sequence homology or domains with other known proteins, but is characterised by a large amount of glutamic acid residues (22%) as well as a clustering of lysine residues on the C terminal region.82 HumanMFAP-1 does not contain any cysteine residues. Interestingly, MFAP-1 was mapped to chromosome 15q15-q21, the same region asFBN1, which madeMFAP-1 an alternative positional candidate gene for MFS. However, extensive mutation analysis failed to show mutations in MFAP-1 in MFS patients, thus ruling out this locus as a reservoir for hidden MFS mutations.83
MFAP2 is an earlier designation forMAGP-1. MFAP3consists of merely two exons encoding a protein of 362 amino acids that displays no homology to other known proteins. The gene is located on chromosome 5q32-q33.2, near to the locus 5q21-q31 reported for the fibrillin-2 gene.84 At present it is not known if the co-localisation of the genes FBN2 andMFAP3 (or of the genesFBN1 and MFAP1 on chromosome 15q15-21) has some functional significance. Interestingly, the gene for the above mentioned lysyl oxidase is located on chromosome 5q23.2-q31.2, whereas a newly discovered lysyl oxidase-like (LOL) gene has been mapped to chromosome 15q24-q25. The function and tissue specificity of the LOL protein remain to be elucidated.85
MFAP4 is a 36 kDa glycoprotein whose tissue distribution appears to be highest in the aortic adventitia.86 MFAP4 has a fibrinogen-like domain and an RGD cell adhesion motif in its N-terminal domain. MFAP4 is one of the genes deleted in the contiguous gene syndrome of Smith-Magenis.87
A family of microfibril-associated proteins termed aortic aneurysm antigenic proteins (AAAP) with distribution limited to the aorta has been identified and preliminary results have been presented.88 89
While the above proteins, and perhaps one or more of the LTBPs, are thought to be integral components of the elastin-associated microfibrils, a number of molecules has been shown to co-localise with these microfibrils or to be able specifically to bind to them (table3).
Molecules which may be able to associate with fibrillin or fibrillin containing microfibrils
A specific binding interaction with fibrillin-1 has been shown for two proteins of the ECM. Fibulin-2 is a four domain ECM protein which is able to form disulphide-linked homodimers.90 Fibulin-2 appears not to be an integral component of the microfibrils, but was shown to co-localise with fibrillin-containing microfibrils in some, but not all tissues.91 In regenerating skin, fibulin-2 associates with fibrillin-containing microfibrils only at late stages of skin morphogenesis.92 Fibulin-2 specifically binds to the N-terminal region of fibrillin-1.91 Since fibulin-2 was also shown to bind with a distinct affinity to fibronectin,93 it seems possible that fibulin-2 plays a connecting role between various ECM proteins, as has been hypothesised for fibulin-1, which can undergo binding with laminin,94nidogen,95 fibrinogen,96 and fibronectin97 as well as self-association.98Interestingly, fibulin-1 was identified within the amorphous core of elastic fibres, similar to elastin, but not in the fibrillin-containing microfibrils.99
An interaction of region C of fibrillin with laminin β2 was identified by yeast two hybrid analysis and confirmed with a protein based affinity assay. Since the laminins are found in basement membranes and fibrillin-containing microfibrils insert into basement membranes, a biological significance for this finding seems possible.100 The laminins are ubiquitous components of the basement membranes and appear to play a role in protein-protein and cell-matrix interactions.101
A series of other proteins has been reported to co-localise or to associate with fibrillin-containing microfibrils. Emilin (elastinmicrofibrilinterface located protein), a 115 kDa glycoprotein, associates with the elastin-microfibril interface. Emilin appears early in embryogenesis and may be involved in elastogenesis.102 103
Fibrillin-containing microfibrils were shown to be closely associated with a chondroitin sulphate proteoglycan; at present neither the nature of the proteoglycan nor whether its protein core or glycose amino glycan chain(s) is responsible for the association with fibrillin is known.104 Interestingly, the expression of decorin may be altered in cases of neonatal Marfan syndrome105 106; as decorin is a member of the small proteoglycan family, it would seem to be a particularly interesting candidate for the proteoglycan association partner.104
Versican, a large aggregating proteoglycan, showed an apparent co-localisation with the elastic network of the dermis in human skin. Since anti-versican antibodies also stained structure resembling microfibrils owing to their oxytolan staining, an association with microfibrils appears very likely.107
Fibrillin polymers as components of the microfibrils
The mechanisms involved in the assembly of fibrillin monomers into polymeric structures have been among the most controversial topics in the field. The key questions include whether fibrillin monomers are arranged in a parallel, head to tail manner or if they form an antiparallel arrangement; whether the monomers are staggered or in register; the sequence of steps involved in polymerisation; the location of self-association sites on fibrillin; if polymerisation occurs by self-assembly or if other proteins or cells are required; what portion of the beads on a string structure of the microfibrils is made up of fibrillin; and how the other components of the microfibrils interact with fibrillin. Although much progress has been made since the discovery of fibrillin in 1986, many of the above questions still remain unanswered.
Taken together, the published evidence speaks strongly for fibrillin monomers polymerising in a parallel, head to tail conformation rather than in an antiparallel fashion. Immunoelectron microscopy with a combination of two monoclonal antibodies shows a periodicity which is only compatible with a head to tail conformation.30 By examination with different monoclonal antibodies it was shown that the N- and C-termini of fibrillin-1 are located in the immediate vicinity and on either side of the beads of the microfibrils, with one bead per fibrillin monomer.30
The fact that individual fibrillin monomers possess a width of 2.2 nm and the microfibrils a width of 10-12 nm23 suggests that the microfibrils may consist of a bundle of individual fibrillin monomers, which is supported by the electron microscopic observation of multiple fibrillin monomers being associated with a single bead.76 108 It remains to ask whether individual, head to tail filaments of fibrillin monomers are arranged in a staggered or an in register fashion within the microfibrils; the evidence reported to date seems to support a combination of these two arrangements.
At least two forms of intermolecular linkage occur in fibrillin microfibrils. Within hours of the extracellular secretion of fibrillin monomers, aggregates are formed which are stabilised by intermolecular disulphide bridges that are not reducible. Fibrillin is thus a covalently bound integral component of the microfibrils.65Probably, multiple fibrillin monomers are associated with a single bead.76 108
Homodimerisation of the N-terminal domain of fibrillin-1 or -2 appears to be the first step in microfibril assembly and is mediated by disulphide bonding.109 The notion of homodimers with an unstaggered alignment is also supported by observations on the tight skin mouse, which carries an in frame duplication of mouseFbn1 exons 17-40, since wild type monomers do not appear to be able to polymerise with mutant monomers of a correspondingly increased length.110
In addition to disulphide bonds, α-glutamyl-ε-lysine crosslinks are also present in fibrillin-containing microfibrils111 and result from modification of fibrillin by transglutaminase.112 The cross links were shown to localise to the interbead regions in a way consistent with a parallel staggered arrangement of fibrillin monomers in the microfibrils.113One way of reconciling the data would be a model in which individual monomers form parallel, unstaggered dimers that in turn associate with other dimers in a staggered fashion.
Whatever the arrangement of fibrillin monomers may be, evidence has underlined the importance of lateral monomer to monomer interactions. FBN1 mutations causing skipping of one or more exons are associated with an almost complete absence of higher molecular weight aggregates in the ECM fraction in cell culture,114 115 which suggests that the correct lateral alignment of fibrillin monomers may be necessary for the assembly of multimeric aggregates. This is compatible with the observation that in frame exon skipping mutations tend to be associated with severe clinical phenotypes.116
Although some issues have been settled concerning the aggregation of fibrillin monomers into multimeric structures, very little is known concerning the interaction of fibrillin with other integral protein components of the microfibrils such as MAPG-1 and -2; to date, it has not been possible to show specific binding interactions.
FBN1 mutations in Marfan syndrome and related type 1 fibrillinopathies
With a few exceptions, all known FBN1mutations identified to date have been unique, which probably reflects the high mutation rate of the fibrillin-1 gene and the impaired reproductive fitness of MFS patients owing to early morbidity and mortality. As of 1998, 137 mutations have been entered in the international Marfan database (http://www.umd.necker.fr) forFBN1 mutations.117 Only 11 of the 137 entries represent recurrent mutations.
The mutations identified to date have been found in almost all exons of the fibrillin-1 gene. Aside from a mild clustering of excess mutations in exons 25, 27, and 28,117 the mutations identified to date appear to be spread across the gene with no clear predilection for any one area of the gene.44 118 119
For reasons that are still not completely understood, mutations have been found in only a minority of patients analysed to date. Following the discovery of the fibrillin-1 gene in 1991, numerous reports on cDNA based mutation analysis of the fibrillin-1 gene in Marfan patients appeared; the sensitivity of mutation detection, when indicated, was between 10 and 25%.120 121 In 1995, the first report on exonwise screening of all 65 FBN1 exons was published; a detection rate of 78% in a group of nine patients was reported.122 However, a subsequent study could duplicate this result only in a small group of well characterised, multigeneration Marfan families (7/9); for the entire group of 60 unrelated patients and families, only 17 probable disease causing mutations were found (28%), suggesting that overdiagnosis of Marfan syndrome may be responsible for the low overall mutation detection rate.123 Other factors, such as the large size of the gene and the large number of exons, may also be partially responsible for the low efficiency of mutation detection in Marfan patients.
The majority of mutations identified to date represent missense mutations (99/137 or 72%); of these, most affect one of the numerous cbEGF modules dispersed throughout the protein (73/137 or 53%). Mutations predicted to lead to premature protein truncation including nonsense mutations and out of frame deletions represent a small proportion of mutations. Exon skipping mutations, associated mainly with mutations affecting splicing consensus sequences, are relatively common (18/137 or 13%). Although mutations of consensus splice sequences are the most common cause of aberrant splicing in human genetic disease, several instances of exon skipping associated with nonsense mutations in the skipped exons have been reported, including one nonsense mutation in FBN1 exon 51.124 125 Unusually, a “silent” mutation in the same exon also was shown to cause skipping of exon 51 in a patient with classical MFS.126
Clear genotype-phenotype correlations have been slow to emerge. In light of the high intrafamilial variability, it is to be assumed that environmental and perhaps epigenetic factors play a significant role in determining the severity of the phenotype in a patient with a given mutation. In addition to mutations found in patients with classical MFS phenotypes, FBN1 mutations have been found in a series of related connective tissue disorders, termed type 1 fibrillinopathies (table 4).
Type 1 fibrillinopathies
Neonatal Marfan syndrome (nMFS) represents the severest end of the spectrum of MFS and is characterised by a series of manifestations, which are rare in classical MFS. Patients with nMFS are typically diagnosed at or shortly after birth and present with symptoms such as mitral or tricuspid insufficiency, congestive heart failure, pulmonary emphysema, joint contractures, crumpled ears, and loose skin. Death occurs usually within the first year of life.127 128 In contrast to classical MFS, where death in untreated patients is most often because of aortic dissection or rupture following progressive aortic root dilatation, the primary cause of death for children with nMFS is severe congestive heart failure owing to mitral and tricuspid regurgitation. If one uses a strict clinical definition of nMFS, there is a clustering of mutations in exons 24-27, where missense mutations as well as small deletions and one exon skipping mutation have been detected in nMFS patients, as well as exon skipping mutations in exons 31-32.127
Patients carrying exon skipping mutations tend to have severe phenotypes,116 and five of the reported nMFS mutations are exon skipping mutations. It appears likely that in frame exon skipping mutations may disrupt the lateral alignment of fibrillin monomers and thereby cause a radical disruption of microfibril formation.116
Autosomal dominant ectopia lentis refers to bilateral ectopia lentis without the typical skeletal and cardiovascular manifestations of the MFS.129 This disease is linked to the fibrillin-1 gene.130 To date, the responsibleFBN1 mutation could be identified in just one affected family.131 The mutation is predicted to substitute the fifth residue of the cbEGF module encoded by exon 59 from glutamic acid to lysine (E2447K). The position within the affected module and the type of residue substitution are not sufficient to explain the phenotype, since a mutation in the cbEGF module encoded by exon 26, which is predicted to substitute the analogous glutamic acid residue to a lysine (E1073K), was found in a patient with nMFS.122
The Shprintzen-Goldberg syndrome (SGS) is a disorder characterised by craniosynostosis and a marfanoid habitus with a range of other features including tall stature, arachnodactyly, dolichocephaly, pectus deformities, scoliosis, joint hypermobility, mental retardation, and more rarely, aortic root dilatation and mitral valve prolapse.132 A de novo FBN1mutation (C1223Y) was found in a patient with sporadic disease,133 and a second change (P1148A) initially reported to be a mutation was later shown to be a polymorphic variant rather than a disease-causing mutation.134 135 It is unknown if mutations in the fibrillin-1 gene are present in most cases of SGS, or if mutations in the fibrillin-1 gene merely contribute to the SGS phenotype in some cases, or whether any true association exists at all.
Autosomal dominant Weill-Marchesani syndrome has been linked to the region on chromosome 15 that containsFBN1.136 Fibrillin immunofluorescence abnormalities were also noted. Patients with Weill-Marchesani syndrome have ectopia lentis, but unlike patients with MFS, are of short stature.
Two FBN1 mutations have been identified in families with marfanoid phenotypes characterised only by skeletal abnormalities. The first (R2726W in exon 64) is predicted to substitute tryptophan for arginine at a site immediately adjacent to a consensus sequence for proteolytic cleavage. The mutation apparently abolished the proteolytic processing of the mutant monomers, which were not incorporated into the ECM. This may provide an explanation for the relatively mild phenotype of the affected family, since the cellular effect of the mutation may thus be equivalent to a “null” allele not expected to exert dominant negative effects.33 The second mutation137 affects a non-conserved residue in the cbEGF module encoded by exon 28 (R1170H) and is of as yet unknown significance. R1170H has recently been described in a second family with skeletal manifestations and mitral valve prolapse.138
The mutation G1127S in FBN1 exon 27 was found in a family in whom ascending aortic disease had been identified in 10 members, nine of whom were shown to carry the mutation. None of them had classical MFS or typical signs such as scoliosis, pectus deformity, arachnodactyly, or ectopia lentis. Although exon 27 encodes a cbEGF module, the residue affected by the mutation G1127S has no known significance for calcium binding.139 NMR analysis of a chemically synthesised peptide consisting of fibrillin cbEGF modules 13 and 14 (exons 27 and 28) with either the wild type sequence or the mutation G1127S showed misfolding restricted to module 13.140 Two further mutations have been reported in patients with isolated thoracic aortic aneurysms,141 but not all families with autosomal dominant inheritance of thoracic aortic aneurysms show linkage toFBN1.142
In summary, the mutation data published to date have shown the existence of a hot spot for nMFS in exons 24-27 and 31-32, and that exon skipping mutations tend to be associated with a severe phenotype. The mutations identified in families with variant phenotypes have not allowed generalisations on the relationships between fibrillin pathogenesis and clinical disease. It is to be hoped that the international databank for FBN1 mutations will contribute to the understanding of FBN1genotype-phenotype correlations in the future.117
Fibrillin-2 mutations in CCA and other phenotypes
Congenital contractural arachnodactyly (CCA) is an autosomal dominant disorder characterised by a marfanoid habitus, arachnodactyly, camptodactyly, crumpled ears, and mild contractures in the elbows, knees, and hips, as well as mild muscle hypoplasia especially of the calf muscles.143 During the quest to identify the genetic basis of MFS, a second fibrillin gene was fortuitously discovered. This second gene was mapped to chromosome 5 (5q23-31) and is now referred to as FBN2. CCA became a good candidate for linkage since its phenotype overlaps that of MFS. Linkage of FBN2 to CCA was indeed confirmed,17 130 and several mutations have been identified in patients with CCA.144-149
Analysis of the nine FBN2 mutations published so far allows for some interesting speculation. All nine mutations are located between exons 24 and 34, analogous to the so called neonatal region of fibrillin-1. It can be hypothesised that this region plays a critical role during development.148
Mosaicism has also been observed in several patients with CCA.145 146 148 In one case, it appears that the fact that the subject was a mosaic helped to minimise the effect of the mutant allele.146 Like the gamut of severity observed in MFS and fibrillin-1 microfibrillopathies, a spectrum of CCA phenotypes also exists. A small number of case reports have described a severe/lethal form of CCA. Molecular studies of one such case has shown that the skipping of exon 34 of FBN2 causes this severe phenotype.146 Comparison of the cardiovascular features between severe/lethal CCA and MFS shows that the changes in the severe form of CCA are consistent with developmental abnormalities, namely septal defects, interrupted aortic arch, and a single umbilical artery. Gastrointestinal abnormalities consisting of duodenal and oesophageal atresia and intestinal malrotation were also seen in some cases of severe/lethal CCA. These developmental abnormalities are consistent with the postulated role of the two fibrillins.
It remains to be seen whether mutations inFBN2 outside the “neonatal region” cause phenotypes that are part of the spectrum of Marfan-like disease. Familial and some sporadic cases of cardiac septal defect, for example, make interesting candidates for continued molecular analysis.
LTBP mutations
Evolutionary comparisons have placed LTBP-2 as the LTBP most closely related to the fibrillins. Therefore, it was hypothesised that mutations in LTBP-2, as well as other components of the extracellular matrix microfibrils, may cause disorders whose phenotypes overlap those of MFS. Since abnormal fibrillin immunofluorescence may be found in disorders caused by defects in both FBN1 andFBN2,16 128 146 the possibility that similar abnormalities would be observed in subjects with LTBP-2 mutations was considered. Preliminary studies have shown at least one putative mutation in a patient and his mother with pectus deformities (Mathews and Godfrey, unpublished data). This amino acid substitution is in a highly conserved region in a cbEGF domain and analogous substitutions inFBN1 have been documented in nMFS and ectopia lentis.
Fibrillin mRNA expression and the dominant negative pathogenesis of Marfan syndrome
mRNAs which carry a premature termination codon (PTC) owing to either a frameshift or a nonsense mutation are usually present in reduced concentration. As a general rule, the closer a PTC codon occurs after the initiation codon, the more likely it is that the mutant mRNA will be reduced in abundance.150 Several PTC mutations in the fibrillin-1 gene have been characterised as having reduced mRNA expression.120 122 151 A 4 bp deletion in exon 41 was identified in a patient with the so called MASS phenotype (Myopia, Mitral valve prolapse, mild Aortic root dilatation without dissection, Skin abnormalities (striae), and Skeletal involvement). The mutant mRNA was expressed at a level of 6% of the normal allele.151In contrast, a mutation leading to skipping of exon 2 (83 bp) with frameshift and PTC was associated with classical MFS; in this case, the mutant mRNA was expressed at a level of 16% of the normal allele.151 The authors concluded that the reduced expression of the mutant peptide leads to a preponderance of normal fibrillin monomers in the polymeric microfibrils, so that below a certain threshold of expression, there is mild disease. On the other hand, if a certain threshold is crossed (between 6 and 16% of wild type levels), the mutant, C-terminally truncated peptide disturbs the polymeric structure to such an extent that more severe disease is seen. It also suggests that the N-terminal region of fibrillin may be crucial for microfibrillar assembly and that it is able to interact in a dominant negative manner with the wild type monomer.151According to the dominant negative model of pathogenesis,152 the product of the mutant allele interferes with the function of the wild type allele. In the case of the MFS, this model seems particularly attractive, because fibrillin monomers aggregate in large, multimeric structures in the course of the formation of microfibrils. Mutant monomers would then disturb the function of the multimeric microfibrils.
Calcium binding and mutations in cbEGF motifs
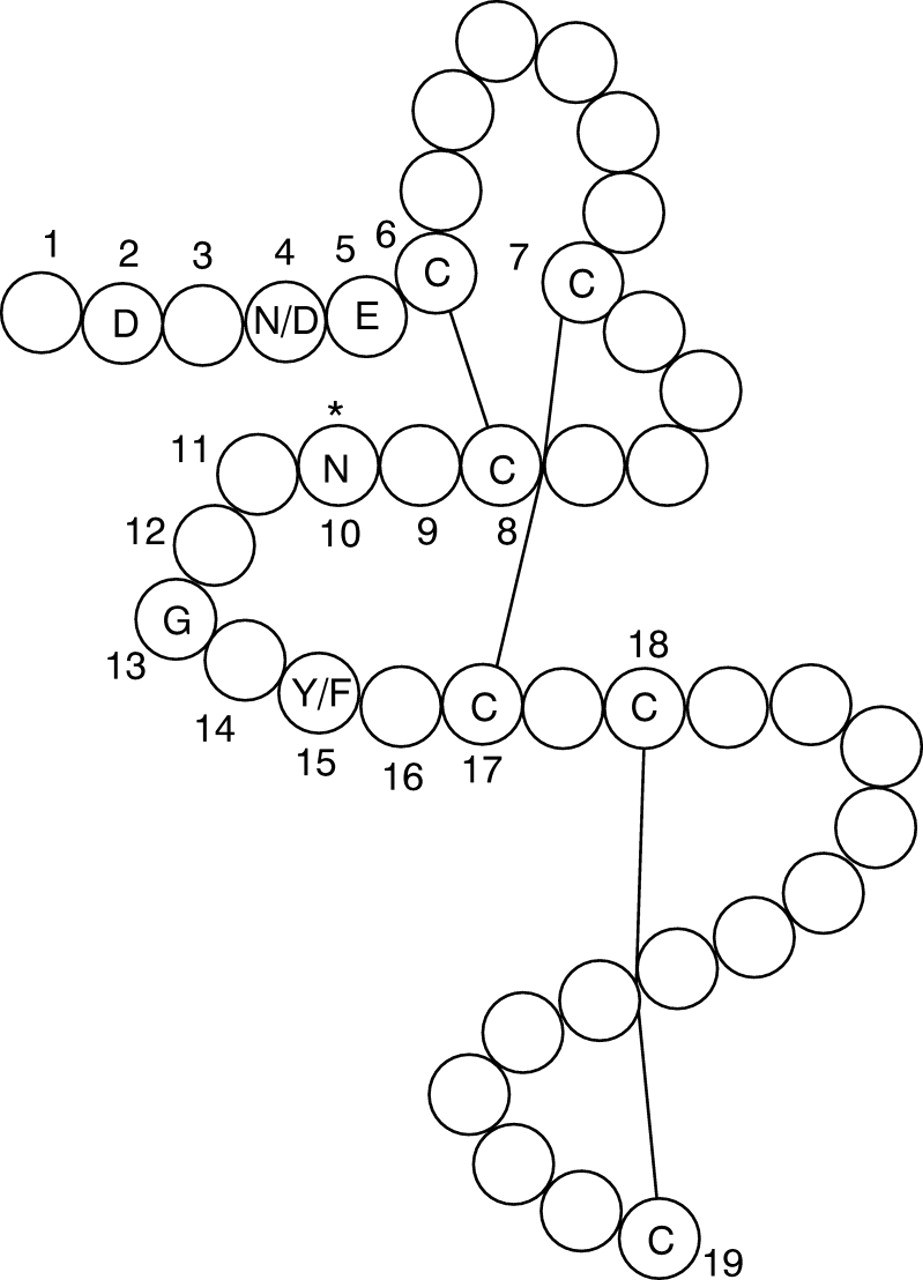
Epidermal growth factor-like (EGF) modules are approximately 45 amino acid residues long and contain six conserved cysteine residues that are paired to form disulphide bonds in a characteristic manner (1 to 3, 2 to 4, and 5 to 6). In addition to the three disulphide bonds, EGF modules are characterised by a double stranded beta sheet as a main structural feature. A subset of EGF modules also contains a consensus sequence for calcium binding (cbEGF), X-D/N-X-D/N-E/Q-C1, comprising the first five residues before the first cysteine residue (C1) in the context of an EGF residue, which also shows a consensus sequence for β-hydroxylation between the third and fourth cysteine residues: C3-X-(D/N)-X-X-X-X-(F/Y)-X-C4.24 153-155In these consensus sequences, X is used to refer to any amino acid, but a non-random distribution of amino acids (three to six at any given position) was found in a sequence alignment of 154 EGF-like domains, which suggests that these positions are important for calcium binding.155 Including the above consensus sequences and the six cysteine residues, 19 of the on average 42 residues of each cbEGF module of fibrillin-1 are predicted to play an important role in calcium binding (fig 2).
{kind=link}
{kind=link}
A calcium binding epidermal growth factor-like motif (cbEGF).The positions marked with the numbers 1 to 19 comprise the six highly conserved cysteine residues as well as the consensus calcium binding sequence, which in turn contains a consensus sequence for β hydroxylation (see text).
Native fibrillin binds calcium,156 but there appear to be significant variations in the calcium binding affinity among different cbEGF modules along the length of fibrillin-1, such that some cbEGF modules may be only partially saturated with calcium under physiological conditions.157 It is conceivable that partially saturated domains may be more flexible than higher affinity domains.157 Calcium coordination is thought to be mediated by a pentagonal, bipyrimidal structure, with the residues of the calcium binding consensus sequence either providing direct oxygen or carbonyl ligands, or playing a role in the stabilisation of the calcium binding site.158 Binding of calcium by one cbEGF module can be influenced by calcium binding to other tandem cbEGF modules, such that the binding affinity is significantly increased.159
Although NMR analysis of a pair of cbEGF modules from fibrillin-1 showed that no ligand is donated from one cbEGF module to the next,160 as is the case with the two cbEGF modules of factor IX,158 calcium binding does appear to rigidify the interdomain region between two cbEGF repeats,160 161which in turn enables multiple tandem cbEGF repeats to take on a rigid, rod-like conformation.162 Removal of calcium by incubation in EDTA leads to a reversible disruption of microfibrillar structure, in which the region between the beads takes on a frayed appearance under electron microscopy.163 This suggests that domains B and D (which contain all of the cbEGF modules of fibrillin-1 and -2) are located in the interbead region. Electron microscopic analysis of a recombinant fibrillin fragment containing a stretch of 12 uninterrupted cbEGF modules found in region D suggested that calcium binding causes the stretch of cbEGF modules to assume a stabilised, more extended, and rigid conformation.164 Tandemly repeated EGF modules are a common motif of proteins of the ECM, such as laminin, thrombospondin, and tenascin, and form extended, rod-like domains essential for the global structure of these proteins.165 In contrast, the interdomain linkage between an N-terminal LTBP module and a C-terminal cbEGF module may be significantly more flexible.166 In addition to the structural functions of EGF modules described above, EGF modules have been shown to mediate calcium dependent protein-protein interactions.167 At present, it is not known if the cbEGF-like modules of the fibrillins are able to mediate protein-protein interactions.
Calcium binding stabilises fibrillin-1 against proteolytic degradation, which emphasises the importance of calcium binding for the structural integrity of fibrillin-1. Similar findings have been observed in other proteins of the ECM, such as fibulin-1 and fibulin-2,168LTBP-1,169 LTBP-2,58 as well as bovine protein C.170 Fibrillin molecules and fibrillin containing microfibrils can be degraded by serine proteinases171 and matrix metalloproteinases (MMP).172 It is conceivable that some fibrillin-1 mutations may increase the proteolytic degradation of microfibrils in tissue, in turn leading to abnormal microfibril morphology and function.
Calcium binding EGF modules (cbEGF) are found in many different proteins, including numerous proteins of the ECM (laminin, thrombospondin, fibrillin-1 and -2, the LTBPs, fibulin-1, and -2, nidogen), blood coagulation factors (factor IX, X, proteins C and S), and proteins involved in specification of cell fate (Drosophila Notch, Delta, Serrate). Disease-causing mutations in cbEGF modules leading to reduced calcium binding have been identified not only in MFS but also in several other disorders including haemophilia B.173
Mutations affecting residues of the calcium binding consensus sequence can reduce calcium affinity, as has been shown experimentally with a chemically synthesised peptide containing a mutation (N2144S) of a residue of the calcium binding consensus sequence of the cbEGF module encoded by exon 52.174 In addition, rotary shadowing of fibrillin-containing microfibrils from the patient carrying the mutation N2144S showed a frayed appearance of the interbead region,174 as was true for another mutation in the same exon (D2127E), which affects a different residue of the calcium binding consensus sequence.114
A classification of cbEGF mutations according to their effects on structure and function has been proposed.160 Mutations of one of the six conserved cysteine residues probably cause domain misfolding, which may also have deleterious effects on the global structure of fibrillin. Alterations of residues of the calcium binding consensus sequence may result in reduced calcium affinity, which may in turn destabilise the interface between two consecutive cbEGF domains. Mutations not affecting cysteine residues or the calcium binding consensus sequence may have effects on intra- or intermolecular interactions; a mutation (G2627R) of the glycine involved in domain-domain packing between two cbEGF modules160 is predicted to alter the pairwise interaction of the adjacent cbEGF modules. Finally, other mutations which do not alter residues with obvious structural relevance may conceivably be involved in protein-protein interactions.160
Immunohistochemical abnormalities of microfibrils in Marfan syndrome
Indirect immunofluorescence with monoclonal antibodies against fibrillin displays a characteristic immunostaining pattern in skin specimens from healthy controls. The dermal-epidermal junction appears as a bright and nearly continuous band parallel to the basement membrane, below which linear strands of fibres descend. The papillary dermis displays a branching meshwork of fibres of various diameters, whereas deeper in the reticular dermis, thicker undulating fibres of various lengths and diameters predominate. Most microfibrillar fibres in this region are associated with elastin. Many, but not all patients with MFS display qualitative and quantitative abnormalities of the dermal microfibrillar network.16Immunofluorescent abnormalities are also present in cardiovascular tissues affected by MFS, such as the aortic valve, the ascending aortic wall, and the mitral valve.176
Immunofluorescent investigations cannot be used as the sole diagnostic test, since patients with other connective tissue disorders can also show abnormalities of the microfibrillar network when examined by this method (see also the hypothesis for the LTBP-2 studies). Additionally, up to 35% of all patients suspected of having MFS may display an apparently normal fibrillin immunostaining pattern in skin specimens.16 Immunostaining in some older unaffected subjects (>70 years) may appear abnormal.
The microfibrillar network of dermal fibroblast cultures can also be investigated by fibrillin immunofluorescence. In most cases, MFS patients show reduced staining intensity indicating a deficient accumulation of reactive fibrous materials. In most MFS patients, there is a reduced staining intensity in both skin samples and dermal fibroblast cultures, but in about 30% of patients, the apparent content of microfibrillar fibres in the skin is normal or near normal, while fibroblast cultures show clearly decreased accumulations of fibrous materials. Rarely, the converse is true, and even more rarely, a patient with classical features of MFS will have apparently normal results for both investigations. These abnormalities cosegregate with the Marfan phenotype within families.177
Therefore, fibrillin immunofluorescence has neither sufficient specificity nor sensitivity to be used as a routine diagnostic tool. It is important to note, however, that normal skin and fibroblast culture fibrillin immunofluorescence patterns are almost never seen in patients with MFS. Thus, normal findings in both assays make the likelihood of MFS very low.
Metabolic labelling of fibrillin: investigation of fibrillin synthesis, secretion, and deposition in the extracellular matrix
A cell culture approach has been developed to investigate the synthesis and assembly of fibrillin in vivo. Owing to its high content of cysteine residues (ca 14%), fibrillin can be labelled with radioactive [35S]cysteine and isolated by immunoprecipitation. Protocols have been developed to separate the cell fraction, the medium, and the ECM fraction, which is defined as the material remaining on the surface of the culture vessel after the harvesting of the other fractions. With this system, information can be gained about the synthesis and secretion of fibrillin as well as on its deposition into the ECM and its aggregation into multimeric structures.178 The biochemical phenotype defined by these parameters is almost always constant among the affected relatives with MFS.179
The glycoprotein fibrillin is synthesised as a 350 kDa propeptide, profibrillin, which is secreted out of the cell within four to eight hours. Profibrillin is cleaved in the extracellular space to fibrillin, which has an apparent molecular weight of 320 kDa.33 The conversion of profibrillin to fibrillin is calcium dependent.115 Fibrillin monomers are then deposited into the ECM where the formation of large, insoluble aggregates takes place. Most of the deposition of fibrillin occurs within the first eight hours after the pulse.179 Fibrillin is assembled into high Mr disulphide bonded aggregates; the formation of aggregates appears to begin in the medium fraction.178
MFS patients show a variety of abnormalities in this system, including decreased synthesis, delayed secretion, and reduced deposition into the ECM; some patients display no abnormalities detectable with this methodology.179
Defined classes of mutation may have specific effects on fibrillin metabolism in this assay. Mutations of cysteine residues were shown to be associated in most cases with delayed secretion of fibrillin presumed to be because of improper protein folding caused by a disruption of disulphide bonding within the affected domain.180 Delayed secretion may in turn be associated with other abnormalities. For instance, a nonsense mutation in the lastFBN1 exon was shown to cause delayed secretion with resultant over-N-glycosylation of fibrillin.37 It is not known, however, if over-N glycosylation per se causes functional impairments.
Using metabolic labelling of dermal fibroblast cultures from patients with MFS, Aoyama et al 181 developed a five group classification of the biochemical phenotype as defined by the parameters of fibrillin synthesis and ECM deposition.181The FBN1 mutation had been identified in 13 of the 55 investigated cell lines, so that hypotheses concerning correlations between genotype and biochemical phenotype could be made.
Groups I and II were characterised by reduced fibrillin synthesis; group II additionally showed impaired ECM deposition. Mutations found in these groups were either nonsense or frameshift mutations and were associated with reduced mRNA expression. The mutations found in group II showed relatively higher levels of mRNA expression (16-25%). Since the ECM deposition was also significantly impaired in group II, it is conceivable that a relatively greater amount of C-terminally truncated fibrillin in class II was able to impair the deposition of the normal fibrillin protein product in a dominant negative manner.
Groups III and IV both showed normal levels of fibrillin synthesis; group III showed a slight impairment of fibrillin ECM deposition, and group IV showed a severe impairment. All the mutations identified in these groups were missense mutations; while the pathogenesis involved in group IV appeared to be related to dominant negative inhibition of microfibril assembly, the pathogenetic mechanisms of group III seemed to be more heterogeneous. Interestingly, cell lines in group V displayed normal fibrillin synthesis, secretion, and ECM deposition. It remains speculative whether functional defects of fibrillin or microfibrils will eventually be shown for this group.
The classification into five groups has a prognostic significance; the incidence of aortic dissection was found to be higher in patients from groups II and IV (with a postulated dominant negative pathogenesis) than in groups I and III.182
Although most cell lines can be classified according to the above scheme, the range of biochemical phenotypes may be more complex than merely five groups. One patient with a missense mutation affecting a cysteine residue in the cbEGF module encoded by exon 14 showed normal synthesis and ECM deposition associated with delayed secretion.183 The patient was mildly affected, in contrast to an earlier observation that patients with delayed secretion tend to have the most severe phenotypes.179
Electron microscopic abnormalities of microfibrils in MFS
Electron microscopic examination of fibrillin-associated microfibrils shows a string of globular structures joined by fine filaments, giving the appearance of “beads on a string”. The string has a diameter of 10 to 12 nm, the beads of up to 22 nm. Strings with up to 60 beads can be seen by electron microscopy. The beads show a variable periodicity which significantly increases when the tissue extract is subjected to tension; the 52 nm periodicity shown in standard biopsy specimens probably represents the period present in relaxed tissue specimens.65
Taken together, there are four types of electron microscopically visible abnormalities of microfibrils isolated from dermal fibroblast cultures from MFS patients: (1) complete or nearly complete lack of microfibrillar assemblies; (2) poorly defined interbead domains with normal periodicity and morphology of the beads; (3) a “frayed” appearance of the interbead domains; and (4) variable interbead periodicity.114 115 184 185
Although no systematic studies have been published, results published to date suggest there may be a loose correlation between electron microscopic morphology and degree of clinical severity in some cases. For instance, two patients with autosomal dominant ectopia lentis without cardiovascular involvement showed abnormalities of class 3 or 4,185 whereas one severely affected MFS patient showed abnormalities of class 1.114
It is important to keep in mind that these electron microscopy abnormalities are true only in secreted, rotary shadowed fibrils. Examination of skin from numerous MFS patients using transmission electron microscopy has shown microfibrils of normal morphology. No quantitative (electron microscopy is a poor tool for quantitation) or qualitative changes have been found (Keene, Godfrey, and Hollister, unpublished data).
Animal models of the Marfan syndrome
Animal models for human disease can be useful to understand the pathogenesis or to explore novel treatment modalities. The first reported naturally occurring model for Marfan syndrome was a cow with skeletal, ocular, and cardiovascular features resembling those seen in the human disease. The phenotype was passed on from an apparently germline mosaic bull to several calves. Light microscopy of the aorta showed typical elastic abnormalities that are seen in the Marfan syndrome.186 187 Both fibrillin immunostaining and metabolic labelling were abnormal and similar to what is seen in human Marfan syndrome.188 Unfortunately, to date no bovineFBN1 mutation has been reported in this herd.
Periera et al 189 have reported gene targeting experiments in a murine model that recapitulates the vascular abnormalities of the human disease. They deleted exons 19 to 24 (which included a portion of the so called neonatal region) ofFBN1. Mice that were heterozygous for the targeted mutation were morphologically and histologically normal and indistinguishable from wild type mice. They lived normal life spans and were fertile. Mice that harboured both targeted alleles (homozygous mutants) had greatly reduced life spans, abnormal fibrillin immunofluorescence and metabolic labelling profiles, and aortic disease similar to that seen in human Marfan syndrome.
The only known naturally occurring fibrillin-1 gene mutation in mice is a tandem duplication of fibrillin-1 in the Tight skin mouse (Tsk).190 This mouse is heterozygous for a genomic duplication of some 30 to 40 kb encompassing exons 17 to 40 ofFBN1. The Tsk/+ mouse has thickened skin and visceral fibrosis that results from an accumulation of extracellular matrix molecules. Thus, the Tsk/+ mouse has become a model for scleroderma, hereditary emphysema, and myocardial hypertrophy. Tsk/Tsk homozygous embryos do not live past day 8 of development. Interestingly, the mutant fibrillin-1 assembles into abnormal microfibrils with increased interbead distances and aggregated clusters,110 not found in wild type mice. There is to date no recognised human equivalent of the Tight skin mouse.
Conclusions
Since the discovery of fibrillin in 1986 by Sakaiet al,15 significant progress has been made in the understanding of fibrillin physiology and pathophysiology. From the identification of the firstFBN1 mutation in 1991 by Dietzet al 20 to the present, a total of 137 FBN1 mutations have been reported to the international database. FBN1 mutations have been shown to be associated not only with Marfan syndrome, but also with a wide range of connective tissue disorders termed type 1 fibrillinopathies. Mutations have also been found in the closely related fibrillin-2 gene in patients with congenital contractural arachnodactyly, and it appears that disease causing mutations will be found in the future in other genes whose products are involved in microfibril metabolism, such as LTBP-2. Therefore, Marfan syndrome is just one of a group of clinical disorders, which may be termed “microfibrillopathies”.
One of the most interesting topics in the field revolves around the pathophysiological mechanisms whereby fibrillin-1 mutations lead to clinical disease. Do most mutations lead to a more or less global disturbance of fibrillin function with differing degrees of severity? Do mutations associated with milder type 1 fibrillinopathies, such as dominant ectopia lentis, affect only certain functions of fibrillin in a specific manner? Do genotype-phenotype correlations exist for classical Marfan syndrome, which could explain the observation that a family history of severe MFS related cardiac disease is associated with an increased risk of aortic complications in families with Marfan syndrome? Answers to these questions will require much more mutation data together with detailed clinical evaluation of the patients and their families. The international FBN1mutation data bank117 should prove useful in this regard. Progress in the understanding of fibrillin pathophysiology obviously is dependent on advances in research on fibrillin and microfibril physiology. Key questions involve the mechanisms surrounding the polymerisation of fibrillin monomers and other proteins into microfibrils and the nature of the interactions between the microfibrils and other components of the extracellular matrix. The differential expression of the various microfibrillar components during development and between different tissues is just beginning to be unravelled.
Gene therapy for patients with Marfan syndrome is not likely to become reality in the near future. Since a dominant negative mechanism of pathogenesis is assumed to be significant for most cases of MFS, gene therapy for MFS will probably involve the down regulation of the mutant allele by one of a number of methods. Since haploinsufficiency or extreme reduction of the mutant allele may also lead to clinical disorders such as the MASS syndrome, it may be desirable to combine down regulation of the mutant allele (or both endogeneous alleles) with the delivery and expression of normal fibrillin under appropriate regulatory control. Initial experiments suggest that at least the first goal may be reachable; approaches using ribozymes with a sequence complementary to fibrillin mRNA led to specific cleavage of fibrillin mRNA with a corresponding drop in expression of fibrillin protein by transfected cells.191 192 Although the ribozymes used in these experiments were targeted to wild type fibrillin-1 sequences, it is also possible to target at least some mutations in a specific manner.193
Marfan syndrome is a clinical diagnosis and will probably remain so for the foreseeable future. The great majority ofFBN1 mutations identified to date are unique, and there is generally no way of predicting the clinical severity or of understanding whatever genotype-phenotype correlation there may be at a molecular level. The full spectrum of the microfibrillopathies remains to be defined. Nevertheless, the enormous progress made in the last decade and the ever growing scientific interest in Marfan syndrome and the extracellular microfibrils leads one to expect that basic research and perhaps also clinical applications will continue to progress by leaps and bounds.
Acknowledgments
P N Robinson would like thank Gerhard Gaedicke, Andreas Kulozik, and Christian Hagemeier for their friendly support. This work was supported by a grant from the Deutsche Forschungsgemeinschaft (Ro 2005/2-1). MG is an Established Investigator of the American Heart Association (funding was contributed in part by the AHA Florida Affiliate).
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
-