Article Text

Abstract
Purpose: To determine a gene locus for a family with a dominantly inherited vestibulocerebellar disorder characterised by early onset, but not congenital nystagmus.
Design: Observational and experimental study.
Methods: We carried out a phenotypic study of a unique four generation family with nystagmus. We performed genetic linkage studies including a genome wide search.
Results: Affected family members developed vestibulocerebellar type nystagmus in the first two years of life. A higher incidence of strabismus was noted in affected members. Haplotype construction and analysis of recombination events linked the disorder to a locus (NYS4) on chromosome 13q31-q33 with a lod score of 6.322 at θ=0 for D13S159 and narrowed the region to a 13.8 cM region between markers D13S1300 and D13S158.
Conclusions: This study suggests that the early onset acquired nystagmus seen in this family is caused by a single gene defect. Identification of the gene may hold the key to understanding pathways for early eye stabilisation and strabismus.
- nystagmus
- familial
- gene
- NYS4
Statistics from Altmetric.com
Pathological nystagmus (involuntary oscillation of the eyes) usually occurs in three settings: (1) onset at birth or in the first few months associated with ocular disease (sensory defect nystagmus); (2) a similar early onset with no detectable underlying pathology (congenital idiopathic nystagmus); and (3) acquired (onset after 6 months) associated with a neurological disorder (neurological nystagmus).1 Although nystagmus is usually sporadic, familial nystagmus can occur usually in the context of sensory defect or neurological nystagmus, when the inheritance pattern reflects that of the underlying ocular or neurological disease. Dominantly inherited pedigrees with isolated nystagmus have been published extremely rarely and usually refer to congenital, rather than acquired nystagmus.2–5
We have identified a unique four generation family with dominantly inherited nystagmus that develops in the first 1 to 2 years of life. The family was initially published several years ago, but the pedigree has enlarged since that time (OMIM 193003).6 Although similar eye movement disorders have been described in dominantly inherited conditions such as the group of spinocerebellar atrophies (SCA), the early onset and lack of progression or other neurological features make these other diagnoses unlikely. The features bear some resemblance to the condition autosomal dominant episodic ataxia, which represents several clinical entities. However, given that only a few of our family members manifested any unsteadiness, this would represent an extremely mild variant of this condition.
Great inroads have been made recently into localising and identifying genes for ataxia and nystagmus. A large number of different genes and loci for SCA has now been identified.7–10 Several genes have now been identified for episodic ataxia, one of which is allelic to SCA6.11–15 Dominant congenital nystagmus has been mapped to a locus on 6p12 (NYS2),4 cosegregating with a balanced translocation 7;15 (t(7;15)(p11.2;q11.2))16 and there is a third type, where 6p12 or 15q11 have been excluded.17
We present the results of a genome wide search on this pedigree, which has resulted in a locus for this disorder on 13q31-q33.
METHODS
After ethical approval was obtained, all available affected and apparently unaffected family members, including spouses, were neuro-ophthalmologically assessed by a single ophthalmologist (NR), orthoptist (JW), and eye movement scientist (CH). Blood was collected and DNA was isolated from 30 family members as indicated on the pedigree. We examined all the subjects concerned who have supplied blood with two exceptions (and by report these subjects were unaffected).
Preliminary linkage studies to regions 6p12 and 7p11.2 were performed and showed that the disease in this pedigree did not link to these loci.
DNA analysis
A genome wide screen, excluding the X chromosome (since male to male transmission was present), was performed on DNA samples using ABI medium density Linkage Mapping Set-MD10 (LMS-MD) (Applied Biosystems). This set consists of fluorescently labelled PCR primer pairs for 400 highly polymorphic dinucleotide repeat microsatellite markers chosen from the Généthon human linkage map.18–20 The markers have an average spacing of 10 cM and incorporate reverse primer tailing chemistry.21 PCR reactions were performed for each marker individually, in a 5 μl reaction volume containing 50 ng of DNA, 2.5 mmol/l MgCl2, 15 mmol/l Tris-HCl, pH 8.0, 50 mmol/l KCl, 250 mmol/l each dNTP, 0.625 pmol of each primer, and 0.25 units of AmpliTaq Gold (Applied Biosystems). Reactions were performed on a Perkin-Elmer 9600 or Hybaid PCR Express thermal cyclers. A standard thermocycling profile was used for all markers consisting of an initial denaturation at 95°C for 12 minutes, which was followed by 10 cycles each of denaturation at 95°C for 15 seconds, annealing at 55°C for 15 seconds, and synthesis at 72°C for 30 seconds. This was followed by 20 cycles each of denaturation at 89°C for 15 seconds, annealing at 55°C for 15 seconds, and synthesis at 72°C for 30 seconds and by a final extension step at 72°C for 10 minutes. PCR products for selected sets of markers were pooled, ethanol precipitated, and size fractionated on a 5% denaturing polyacrylamide gel (Amresco) by electrophoresis on an ABI 377XL sequencer. PCR products were sized by the GeneScan version 3.1 program, and were scored using the Genotyper version 2.5 program. In addition to the 400 markers from the LMS-MD10 set, a further 17 markers were used in genotyping from LMS-HD5 and the Généthon microsatellite linkage map20 (fig 1). PCR reaction and thermocycling conditions were similar to those used for the LMS-MD10 marker set, but different annealing temperatures were used for each primer pair after optimisation using a Hybaid PCR Express gradient thermal cycler.
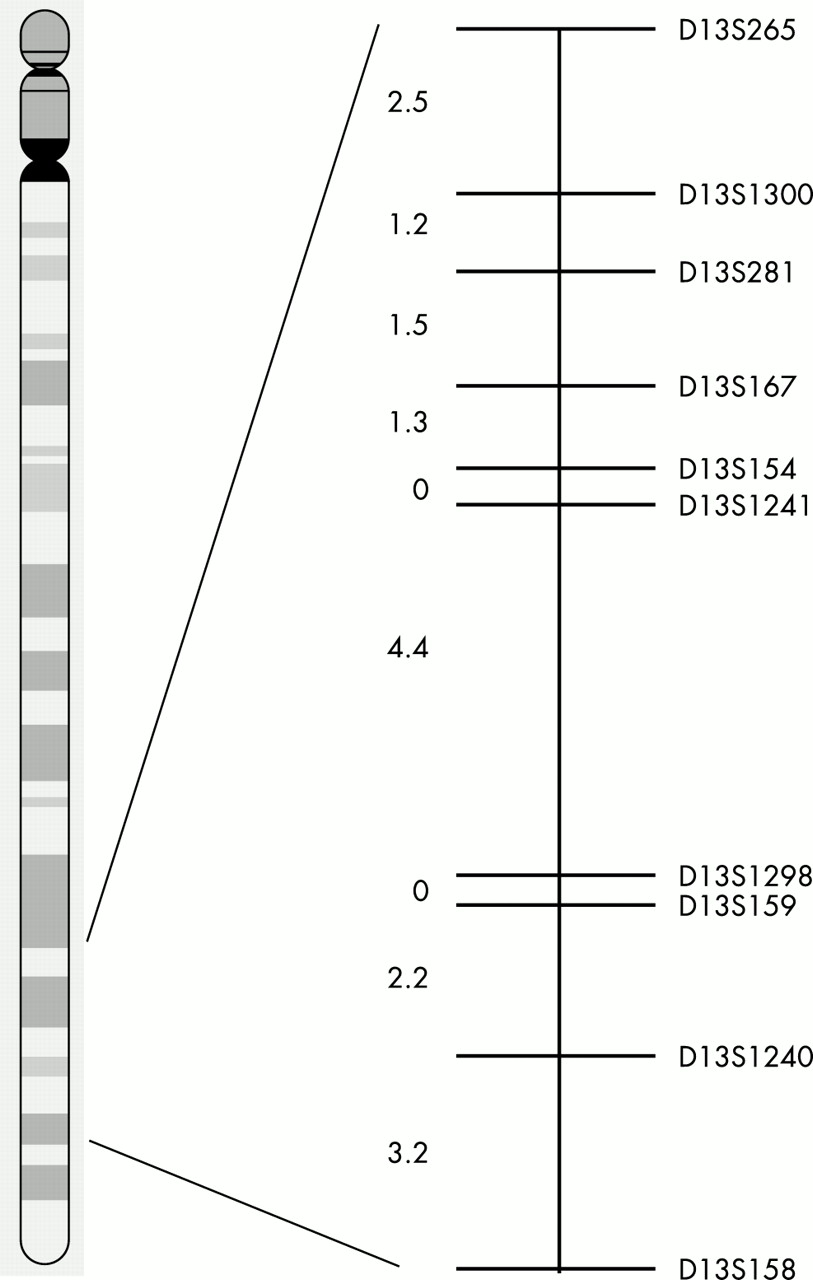
Schematic genetic map showing marker order and genetic distances (cM) between the chromosome 13q31-q33 region. Distances from the Généthon microsatellite linkage map.20
Linkage analysis
Microsatellite data were checked for genotyping errors using the PEDCHECK.22 Pairwise linkage analysis was performed by the MLINK program of the FASTLINK version 4.0P package,23 accessed via the Genetic Linkage User Environment interface (UK HGMP Resource Centre). For linkage calculations, allele frequencies were assumed to be equal, the gene frequency for nystagmus was assumed to be 1 in 5000 with complete penetrance, and male and female recombination rates were assumed to be equal.
RESULTS
The clinical features of this family have been previously described6 and are further summarised in table 1. In brief, the affected subjects nearly all acquired an eye movement disorder within the first two years of life. This timing is usually of concern as it may indicate intracranial disease. In some family members there was associated mild dizziness or loss of balance. The disorder was not characterised by paroxysmal exacerbations or progressive deterioration. No abnormalities have been shown on neuroimaging.
Summary of clinical findings in family members
Vision was very well preserved in most subjects, except those with strabismus related amblyopia. Strabismus was strongly associated with this condition, being present in nine out of 14 affected members (10 out of 14 if II.10 is included), but only one out of 10 unaffecteds (two out of 10 if IV.37 is included who had an inconsequential exophoria). Exophoria is commonly present in normal subjects. The twins (III.25 and III.26) had profound hearing loss necessitating hearing aids. This may have been related to the severe neonatal problems that they both experienced. The mild balance problems were not typical of affected family members and were therefore assumed to be unrelated. The eye movement abnormalities were characterised by poor or absent smooth pursuit, gaze evoked nystagmus, upbeat nystagmus, and poor vestibulo-ocular reflex.
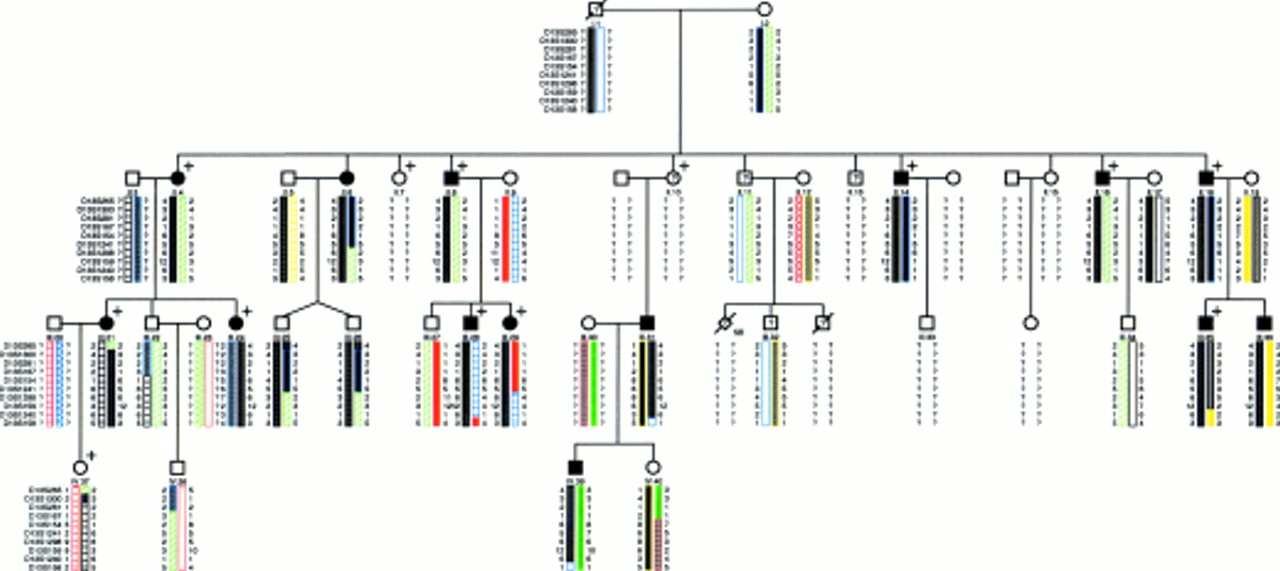
The pedigree is shown in fig 2, indicating the family members that were affected clinically, those with strabismus, and the results of the DNA analysis. The proband, III.28, has neurofibromatosis type 1, inherited from his mother II.9. The linkage analysis for nystagmus is shown in table 2. A maximum lod score of 6.322 at θ=0 for D13S159 was shown for nystagmus for markers in the 13q31-q33 region.
Lod scores for nystagmus with markers in the chromosome 13q31–q33 region
{kind=link}
{kind=link}
Pedigree showing haplotypes for markers in the chromosome 13q31-q33 region. Recombination events in affected subjects narrow the critical region to a 13.8 cM region between D13S1300 and D13S158. Distances from the Généthon microsatellite linkage map.20 Filled symbols = affected subjects. Unfilled symbols = unaffected subjects. ? = Unknown. This category includes: (1) subjects who were not examined (I.1 (dead), II.7, II.13); (2) subjects who were not examined, but supplied blood (II.11 and III.32); (3) subjects who were unassigned as signs or symptoms were inconclusive (II.10). SB = stillborn. + = affected with strabismus.
DISCUSSION
This unique familial disorder is characterised by an onset of gaze evoked nystagmus and associated eye movement abnormalities in the first two years of life with no or minimal ataxia or other neurological signs. The disorder appears to be non-progressive; the oldest affected member is now 48 years old. As described previously, the type of nystagmus most resembles that seen in acquired vestibulocerebellar disorders, rather than congenital or latent types.6 We previously proposed either a novel condition or an unusually benign variant of one of the dominant ataxias.6 Our results now confirm the existence of a locus on chromosome 13q31-q33 that does not correspond to any known existing gene or locus responsible for either congenital nystagmus or the cerebellar ataxias.
Although the overall phenotype in this disorder is very unusual, the oculomotor signs by themselves are relatively common. Gaze evoked nystagmus and poor/absent smooth pursuit can be seen in a wide range of progressive cerebellar disorders and may also be present in stationary cerebellar malformations. A variety of drug toxicities may also lead to the same abnormality (alcohol, anticonvulsants). In animals, identical signs can be induced by lesions in the flocculus and paraflocculus of the vestibulocerebellum, which has led to the term “flocculus syndrome” to describe these eye movement abnormalities.24 It is widely believed that one of the functional roles of the flocculus is to maintain stability of the retinal images of stationary or moving visual objects by adaptive control of eye movements. Thus, the failure of this function leads to poor gaze holding when the eyes are eccentric from their primary position (leaky integrator) leading to gaze evoked nystagmus and an inability to generate the oculomotor commands to follow a moving object smoothly (poor or absent smooth pursuit).
The incidence of strabismus was much higher in the affected than in the unaffected family members and considerably higher than in the general population. This strong linkage of strabismus to the eye movement disorder in this pedigree is very interesting. Both cerebellar flocculus and vermis neurones are involved in the control of vergence.24 One possibility, therefore, is that there is a primary abnormality affecting the vergence system, although we are not aware of strabismus resulting from floccular lesions. Alternatively, the strabismus may be a non-specific result of relatively early onset oculomotor dysfunction.
Genes involved in CNS development or maintenance might be considered candidate genes. Potential candidate genes in this region might include SOX21, ZIC2, and TYRP2 and work is in progress to investigate this possibility. SOX21 (SRY-box21) (MIM 604974), a member of the SOX family of developmental genes, is expressed in human embryonic brain.25ZIC2 (ZIC family member 2) (MIM 603073) is a developmental gene that is known to give rise to holoprosencephaly if there is haploinsufficiency or genetic mutation.26 Although, our family does not show brain abnormalities, it is possible that the same gene may have a more minor mutation. TYRP2 (MIM 191275) dopachrome tautomerase is a tyrosinase related protein, important in the correct development of pigmentation, including choroidal pigmentation. Generalised and ocular albinism is associated with nystagmus, although not of the exact type seen here. Rather than it being a prime candidate, it is important to exclude.27
In this family, at present we have no anatomical correlates for their eye movement abnormalities, and can only conclude that there is a failure of floccular function, either through an abnormality of the flocculus itself, or through other structures that project to the flocculus. Regardless, it is striking that this function can be singled out and linked to a single genetic locus, leading us to surmise the existence of a gene critical to establishing early eye movement control pathways.
Acknowledgments
The study was performed at the Department of Ophthalmology, Great Ormond St Hospital for Children, London, and the Medical Research Council HGMP Resource Centre, Cambridge. We are grateful to the family involved in the study for their participation. We thank Anna Glaser PhD in Professor P Scambler’s laboratory, Institute for Child Health for performing the initial linkage studies. The genome screen was performed at the Medical Research Council UK HGMP Resource Centre Linkage Hotel. We thank the Ormsby Foundation for their financial support.
Electronic database information. Accession numbers and URLs for data in this article are as follows: Genome Database, http://www.gdb.org. Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim (for (MIM 193003)). UK HGMP Resource Centre, http://www.hgmp.mrc.ac.uk (for GLUE interface and other linkage utilities).