Article Text

Abstract
Aims and background: Mutations in the TARDBP gene, which encodes the TAR DNA binding protein (TDP-43), have been described in individuals with familial and sporadic amyotrophic lateral sclerosis (ALS). We screened the TARDBP gene in 285 French sporadic ALS patients to assess the frequency of TARDBP mutations in ALS.
Results: Six individuals had potentially deleterious mutations of which three were novel including a Y374X truncating mutation and P363A and A382P missense mutations. This suggests that TARDBP mutations may predispose to ALS in approximately 2% of the individuals followed in this study.
Conclusion: Our findings, combined with those from other collections, brings the total number of mutations in unrelated ALS patients to 17, further suggesting that mutations in the TARDBP gene have an important role in the pathogenesis of ALS.
Statistics from Altmetric.com
Amyotrophic lateral sclerosis (ALS, MIM 105400) is a devastating neurodegenerative disorder of incompletely understood aetiology which is characterised by degeneration of neurons involved in motor neurons from the spinal cord, brainstem and neocortex.1 Most affected individuals present with progressive paralysis and, in general, death is caused by respiratory failure within 3–5 years after symptom onset. Approximately 10% of ALS cases are familial whereas the remaining 90% are sporadic.2 Mutations in the Cu/Zn superoxide dismutase 1 (SOD1) gene account for approximately 12–20% of familial ALS cases.3 However, mutations in other genes have since been reported for ALS, including VAPB, ALS2, SETX, DCTN1 and ANG, which together account for only a small proportion of ALS cases.4
The presence of ubiquitinated neuronal cytoplasmic inclusions (NCI) is considered one of the pathological hallmarks of many neurodegenerative diseases including ALS.5 TDP-43 has been identified as a major constituent of τ-negative ubiquitin positive NCIs.6 7 Several disorders including frontotemporal lobar dementia (FTLD) as well as ALS are now referred to as TDP-43 proteinopathies.8 Moreover, the observation of prominent abnormal molecular weight fragments of TDP-43 in affected nervous tissues of patients strongly suggests a causative role in neurodegeneration.7
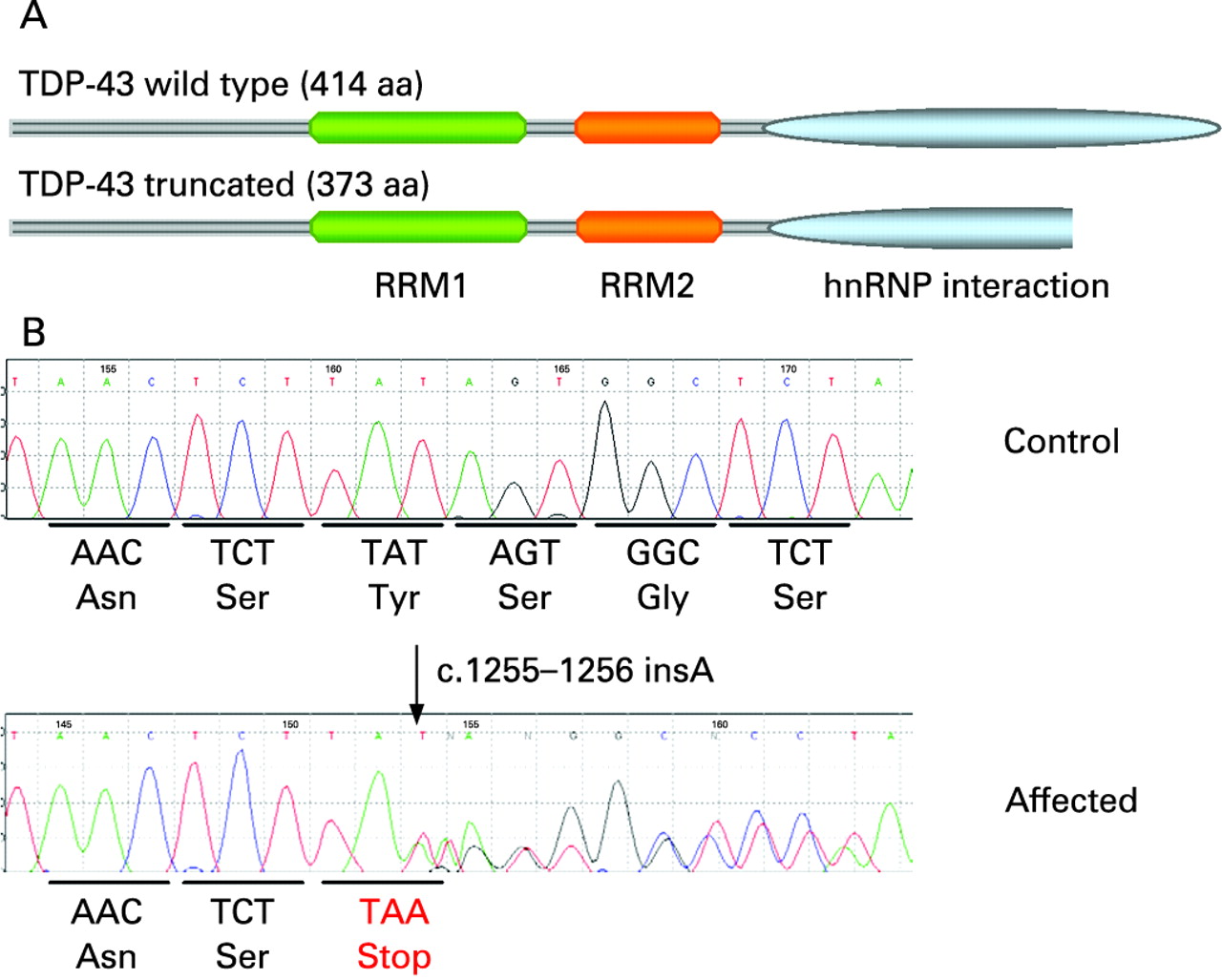
TDP-43 is an evolutionarily conserved 414 amino acid nuclear protein which contains two RNA recognition motifs (RRM1 and RRM2) and a glycine-rich C terminal domain.9 The C terminal region of TDP-43 appears to be involved in protein–protein interactions, most notably with hnRNPs.10 The protein is involved in several processes including regulation of gene expression and splicing as well as exon skipping.11 Recently, mutations in the TARDBP gene at the ALS10 locus on chromosome 1p36.2 have been independently reported by our group and others in familial and sporadic patients with ALS.12–16 Two additional studies failed to detect mutations or copy number variants in TARDBP in Belgian and North American patients with ALS.17 18 The 14 reported mutations are all missense mutations that occur in a highly conserved region of the C terminus of TDP-43, except one missense (D169G) arising at the first RNA recognition motif.13
Given the increasing amount of genetic and functional reports that demonstrate the involvement of TDP-43 in the physiopathology of ALS, we sought to assess further the types and frequency of mutations using an extended cohort of sporadic ALS patients from France. We therefore screened 285 new sporadic ALS cases for mutations in TARDBP gene by DNA sequencing.
PATIENTS AND METHODS
Patient and control population
All ALS patients enrolled in this study, as well as unaffected individuals, were recruited through clinics in France and independently ascertained by no less than two neurologists. All samples were collected with the approval of the relevant institutional ethic boards and the informed written consent from each participant. DNA was extracted from peripheral blood samples using standard methods. The case cohort used in this study consisted of 285 French subjects diagnosed with probable or definite sporadic ALS as per El Escorial criteria.19 Of these, 175 were men and 110 were woman and the average age of symptom onset was ∼59 years; 189 patients had spinal onset disease whereas 96 presented bulbar onset disease. The control cohort used in this study comprised 360 unrelated neurologically healthy individuals who had been previously sequenced for TARDBP.13
Gene screening and variation analysis
The coding region of all five coding exons of TARDBP (accession number NM_007375) was sequenced in each individual and the PCR product contained a minimum of 50 bp from each of the flanking introns. Oligonucleotide primers were designed using the Exon Primer software from the UCSC genome browser (http://genome.ucsc.edu/cgi-bin/hgGateway) and have been previously published.13 Polymerase chain reactions (PCRs) were performed using the AmpliTaq Gold DNA Polymerase (Applied Biosystems, Foster City, California, USA) as per manufacturer’s instructions. PCR products were sequenced at the Genome Quebec Innovation Centre (Montréal, Québec, Canada) using a 3730XL DNA analyser. In each case, the forward primer was used for sequencing and variations were confirmed by reverse sequencing. Mutation surveyor software (version 3.10) was used for mutation detection analyses (SoftGenetics, Pennsylvania, USA).
Bioinformatics
Two software programs were used to predict the overall severity of the identified mutations: PolyPhen (http://genetics.bwh.harvard.edu/pph/)20 and SIFT (Sorting Intolerant From Tolerant).21 Default conditions were used for the two programs.
RESULTS
Following the DNA sequencing of the entire coding sequence and splice junctions of TARDBP, nine individuals had a total of six distinct variants, four of which had not been previously described (table 1, supplemental fig 1).
{kind=link}
Of the nine individuals, six had mutations that are considered pathogenic. One variant was particularly interesting and novel as it was the insertion of an adenine in exon 6 (c.1255-1256insA) that creates a premature stop codon (p.Y374X) and is predicted to produce a truncated TDP-43 protein (fig 1). To our knowledge, this is the first description of a truncating mutation in the TARDBP gene in a patient with ALS. Two additional previously unreported heterozygous missense mutations (P363A and A382P) were identified in two patients, as well as a G348C missense in three unrelated patients which had already been reported also in a French patient.13 The P363A mutation is conserved in five of eight species examined; however, regarding the A382P missense, both the mouse and rat have a praline at this position (supplemental fig 2). Moreover, we identified a variant in the 3′UTR region (c.1462T>C) of another patient that was not observed in 360 controls, which may have an impact on the expression or regulation of TDP-43; however, we cannot conclude that it is pathogenic. Finally, one silent mutation was found (p.A66A) in two unrelated patients but is likely to be a benign polymorphism as it is predicted not to affect function.
DISCUSSION
In this study we undertook a mutational analysis of the TARDBP gene to evaluate the types and frequency of mutations in our French cohort of sporadic ALS cases. We identified a frameshift mutation which creates a premature stop codon (p.Y374X) that will consequently lead to the expression of a truncated protein. This mutation was absent from 360 French controls and from other published control and patient screens of TARDBP. It removes the last 41 amino acids of the protein which can be expected to alter or abrogate the interaction of the TDP-43 C terminal region with hnRNPs A/B and A1a.10 While the impact of such a truncation of TDP-43 will need to be evaluated, it is quite plausible that the truncation could affect TDP-43’s role in gene expression as well as exon splicing.11 Moreover, this truncated form of TDP-43 may acquire novel toxic functions through established or unknown abnormal interactions; alternatively it may also increase the aggregation propensity of the protein and its recruitment in NCI. Of note, small truncating mutations have also been identified in the SOD1 gene, including a L126X mutation that removes the last 28 amino acids of SOD1.22
Key points
From a cohort of 285 French sporadic amyotrophic lateral sclerosis (ALS) patients, six presented potentially deleterious TARDBP mutations of which three were novel, including an Y374X truncating mutation and P363A and A382P missense mutations.
This is the first report of a truncating mutation in the TARDBP gene occurring in a patient with ALS.
These findings combined with others further support an important role for TDP-43 in the pathogenesis of ALS.
We identified three heterozygous missense mutations in exon 6 of TARDBP (P363A and A382P and G348C) in five unrelated patients. This seems to represent a hotspot for ALS linked TARDPB mutations. In addition, the identification of the same G348C missense mutation in three patients but no controls suggests that it is particularly damaging. Finally, we identified a variant in the 3′UTR region (c.1462T>C) in another patient which may be pathogenic given that it was not observed in 360 controls. Unfortunately, it was not possible to conduct experiments to test the effect of the mutations on the TDP-43 protein in individuals with the (c.1255-1256insA) and (c.1462T>C) variants as lymphoblastoid cell lines were not available.
The number of mutations in patients predicted to be pathogenic that we detected in this study (6 of 285; 2.1%) is comparable to other studies. Our previously reported results for TARDBP mutations in French and French Canadian ALS patients were 6 in 120 sporadic patients (5%) and 3 in 80 familial patients (3.8%).13 Combining the number of missense changes with the previous results in sporadic patients yields a rate of 12 in 405 (2.9%).
In conclusion, we identified for the first time a nonsense mutation in the TARDBP gene in an ALS patient. We also observed two novel missense changes as well as one new 3′UTR variation, therefore enlarging the spectrum of mutations that link TDP-43 neuropathology to ALS neurodegeneration. Our data, combined with the literature, provide evidence that mutations in the TARDBP gene are not rare in sporadic ALS patients. Future investigations will be necessary to establish if it is one of the normal properties of wild type TDP-43 which is lost or altered upon its mutation, possibly through abnormal interactions with RNA and protein. This will help us to understand better the aetiology of ALS and the characteristic degeneration of motor neurons.
Acknowledgments
We would like to thank all the families involved in this study.
REFERENCES
Supplementary materials
web only appendices 46/2/112
Files in this Data Supplement:
Footnotes
Additional figures are published online only at http://jmg.bmj.com/content/vol46/issue2
Funding: GAR is funded by the Canadian Institutes of Health Research (CIHR), Muscular Dystrophy Association USA and ALS Association (ALSA), EK is funded by ALS Canada and CIHR, ND by CIHR, PNV by the Fonds de Recherche en Sante Quebec (FRSQ) and VM by the Association Francaise contre les Myopathies France (AMF) and the Association pour la Recherche sur la Sclerose Laterale Amyotrophique (ARS).
Competing interests: None.
Patient consent: Obtained.