Article Text

Statistics from Altmetric.com
Human bone marrow is a fascinating and complex organ. Its major function is to produce and sustain normal haemopoiesis, and in a normal adult this entails the production of more than 1011cells/day.
The marrow contains most of the multipotent haemopoietic stem cells, and provides an environment for these cells to differentiate into each of the well recognised peripheral blood cells (erythroctes, leucocytes, and platelets). In addition, the marrow nurtures the development of B lymphocytes and provides T cell progenitors which migrate to the thymus and differentiate there into mature T cells.
This brief review is selective and focuses on the neurological aspects of several bone marrow disorders—namely, multiple myeloma, paraproteinaemia, Waldenstrom’s macroglobulin, cryoglobulinaemia, and lymphoma. Neurological aspects of bone marrow transplantation and chemotherapy are also considered.
Multiple myeloma
Multiple myeloma is the most common of the plasma cell dyscrasias, which also include monoclonal gammopathies of unknown significance (MGUS or paraproteinaemias), plasmacytomas, and plasma cell leukaemia. These terms represent a range of diseases characterised by a monoclonal proliferation of plasma cells, and associated with a corresponding diversity in clinical behaviour.
Antibody molecules, the product of plasma cells, are composed of two heavy and two light chains. There are five heavy chain isotypes (G, M, A, D, and E) and two light chain isotypes (κ and λ). Each chain consists of a constant region and a variable region. The second provides the recognition site for the antibody molecule; its structural uniqueness or idiotype derives from a particular clone of cells as each antibody is produced by a single clone. Serum electrophoresis produces a broad peak in the γ region, which is composed of a very large number of immunoglobulin molecules each specified by a unique plasma cell clone. In patients with multiple myeloma and other plasma cell dyscrasias a sharp spike or monoclonal (M) component is found within the γ region; this M protein represents a single protein, the product of a single clone of cells. Immunoelectrophoresis may be used to show that the M component is composed of a single light and heavy chain type—that is, it is truly monoclonal. The amount of M protein in a given patient provides a reliable measure of tumour burden and is thus a useful measurement. Bence Jones proteins, which represent free light chains, are produced in excess and excreted in the urine in 50% of cases of multiple myeloma.
Multiple myeloma is a relatively rare cancer, which occurs predominantly in patients over 60 years of age, although a few patients can present in their 20s and 30s. The disease is characterised by a triad of features; lytic bone lesions, the production of a monoclonal paraprotein, and an increase in abnormal plasma cells in the bone marrow. Associated features may be bone pain, renal impairment, anaemia, and hypercalcaemia. Even with modern treatment regimens, including bone marrow transplantation, the disease is essentially incurable, with median survival being about three years, although some patients will succumb very quickly and a few will survive beyond 10 years.
Patients with multiple myeloma are commonly referred for neurological opinion because of frequent involvement of the nervous system.
INVOLVEMENT OF NERVE ROOTS AND SPINAL CORD
The commonest presenting symptom of multiple myeloma is bone pain. It has been reported in 80% of cases and in 60% it is located in the back, most commonly in the lumbosacral region (fig 1).1Radicular pain afflicts one in five patients. Compression of the spinal cord or nerve roots also affects about 20% of cases.1 2The thoracic region of the spinal cord is involved more often than the other areas and patients with IgA myeloma seem to be at greater risk of spinal cord compression. Spinal cord compression with consequent neurological emergency results from vertebral collapse or from tumour mass deriving from vertebrae or an epidural site (fig 2). Solitary plasmacytomas may occasionally compress the spinal cord or even a nerve root in which case severe root pain results with radiographic signs similar to acute disc prolapse.

Spinal MRI showing compression collapse of the vertebral body of T11 and a lytic lesion within T12. The patient complained of lower back pain.

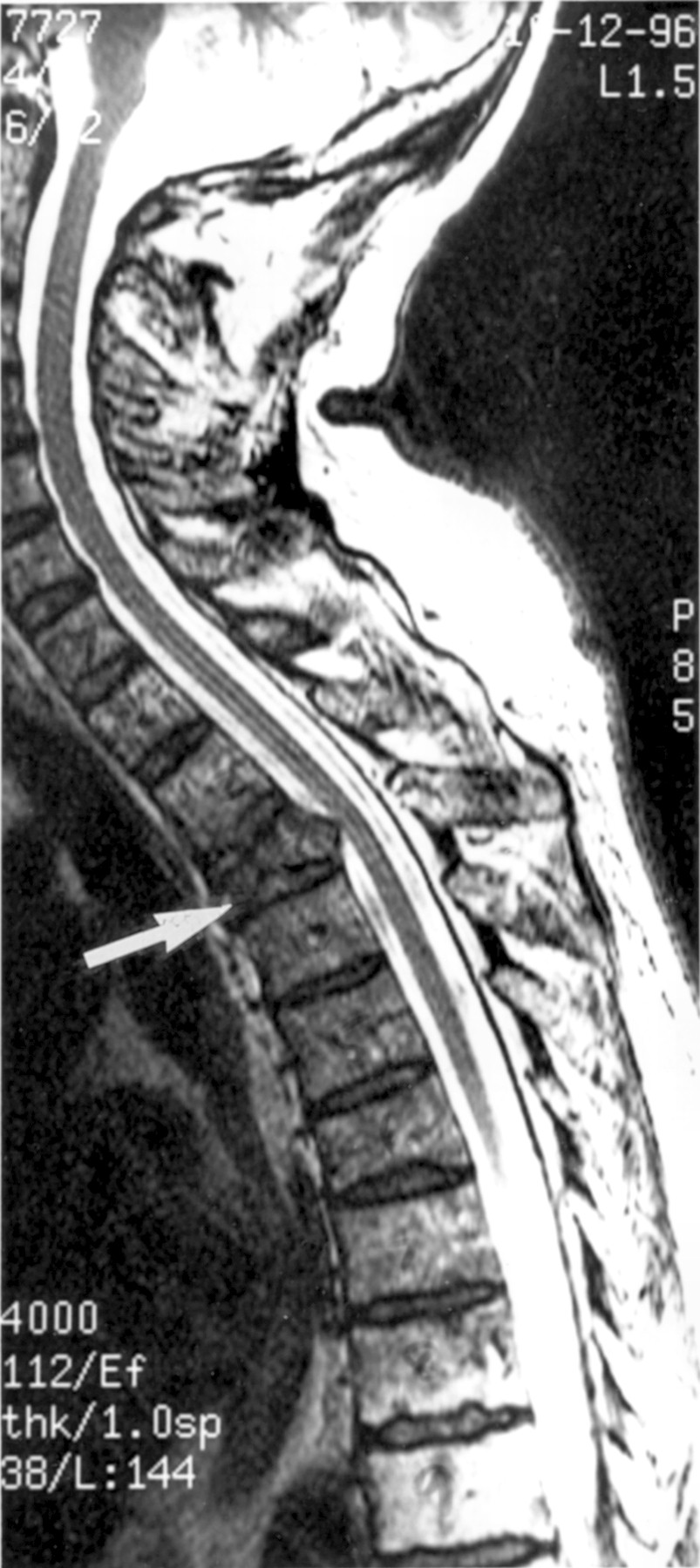
Spinal cord MRI of a patient with multiple myeloma showing compression of the cord after collapse of the third thoracic vertebral body (arrow).
Management of cord compression requires immediate diagnosis so that appropriate treatment may be given before permanent motor or sensory deficits result. To detect early evidence of spinal cord compression and to define the area of involvement neurological assessment should be followed by emergency radiography (MRI). Immediate radiotherapy with steroid cover is usually appropriate therapy and decompressive surgery is rarely required,3 except when the diagnosis is uncertain. Local control of disease can usually be obtained with a tumour dose of 2000 cGy given over five days from a 60Co source.4
INTRACRANIAL AND CRANIAL NERVE LESIONS
Although the skull bones are commonly affected by myeloma, involvement of the brain is relatively rare. It may, however, result from lesions within the bony vault or by metastases in cases of plasma cell leukaemia. Rarely will the brain or its coverings be the site of solitary myeloma.5 The clinical features of intracranial myeloma depend on the site of involvement but may include the symptoms and signs of increased intracranial pressure, papilloedema, impairment of consciousness, and focal neurological deficit including cranial nerve lesions.
Cranial nerve signs may result from the involvement of cranial nerve foramina in myelomatous skull base lesions, or from compression or distortion of nerves by tumour masses arising from the sphenoid or petrous bones.5 The most often involved cranial nerves are VI, II, V, VII, and VIII.
The diagnosis, if not apparent from the radiology, is facilitated by the examination of the CSF as all patients with meningeal involvement show plasma cells within the CSF and the CSF protein is usually greatly increased.4 When meningeal involvement has resulted in impaired consciousness or cranial nerve signs, the outlook is poor but intrathecal drug therapy may prove beneficial.3
PERIPHERAL NEUROPATHY
Neuropathy in myeloma is uncommon with a prevalence in retrospective series of less than 5%1 6 but Walsh7 found clinical evidence of neuropathy in 13% in a prospective study, and electrophysiological evidence in 39%.
The neuropathy is most commonly sensorimotor in type but purely sensory or relapsing and remitting forms occur.8 9 Neuropathy of gradual onset is usual but it may be acute or subacute.10Cranial nerves may occasionally be affected and upper limbs more than lower. Some patients have systemic amyloidosis and Kyle and Dyck9 have emphasised the importance of examining appropriate biopsy tissue as this complication may be the cause of neuropathy. The diagnosis may be suspected by the finding of postural hypotension, impotence, pain, carpal tunnel syndrome, or dissociated sensory loss.8
Nerve conduction studies and nerve biopsy most commonly show changes consistent with axonal degeneration although demyelination has been occasionally reported.8 Infiltration of nerve with plasma cells or amyloid may be found.11
Neuropathy in osteosclerotic myeloma (POEMS syndrome)
Unlike the rarity of neuropathy in the common osteolytic variety of myeloma, about half the patients with osteosclerotic myeloma (2% of cases) present with neuropathy and do so at any earlier age. The neuropathy is mainly motor and similar to those of chronic inflammatory demyelinating polyneuropathy (CIDP); it may be part of a syndrome designated by the acronym POEMS12 (polyneuropathy, organomegally, endocrinopathy, M protein, and skin changes). POEMS syndrome is usually seen in the purely sclerotic or mixed sclerotic and lytic types of myeloma, but may occur in the lytic form, in patients with M protein alone (without myeloma), polyclonal protein alone, and rarely in patients with extramedullary plasmacytomas.13
CLINICAL FEATURES
Men are affected more than women (2:1), the average age of onset being 47 and one in five cases are younger than 40.13 14Patients usually present with neuropathy, symptoms of which may precede the diagnosis by one to two years. The neuropathy is usually a dominantly motor, sensorimotor neuropathy, which commences distally and symmetrically and progresses slowly with proximal spread. Severe weakness results with more than half the patients being unable to walk.13 Cranial nerves are not affected but papilloedema may occur. Peripheral (pitting) oedema and hyperpigmentation are common features as are hypertrichosis (the development of stiff black hair on the extremities) and skin thickening. Hepatomegaly is a frequent finding but splenomegaly and lymphadenopathy less so. Gynaecomastia, amenorrhoea, impotence, and testicular atrophy occur, finger clubbing and white nails may be seen in about 50%, and ascites and pleural effusion are occasionally seen. Weight loss occurs early and patients are often wasted and appear cachectic. Facial lipoatrophy may be striking.15 Haemangiomatous proliferation of small blood vessels in the skin, kidney, brain, and lymph nodes has been reported.
Nerve conduction studies show moderate to pronounced slowing of conduction velocity, and the changes within sural nerve biopsy are of mixed segmental demyelination and axonal degeneration.9 12 16 17
Other laboratory findings are unlike myeloma; anaemia is rare and polycythaemia may be present.16 Hypercalcaemia and renal insufficiency are uncommon, the bone marrow is rarely infiltrated with plasma cells, and M components are of modest amount in the serum and rarely present in urine.18 The M protein is usually of the IgG or IgA heavy chain class and of the λ light chain type.14
Tests of endocrine function have shown diabetes mellitus to be present in 50% of patients19 and evidence of thyroid, adrenal, or gonadal failure less commonly. Low serum testosterone is common and increased oestrogen has been reported in impotent men.16
Radiographic skeletal survey is most important diagnostically, particularly as 25% of these patients show no serum or urine protein. Osteosclerotic lesions, or a mixture of sclerotic and lytic lesions, are usually seen, or a sclerotic rim around a lytic lesion.
Lymph node biopsy in some patients has shown the features of Castleman’s disease or angiofollicular lymph node hyperplasia.17 20 21
PATHOGENESIS OF NEUROPATHY
The cause of neuropathy in patients with myeloma remains unclear. Binding of immunoglobulin to neural components has been described in only some cases.22 23 Reactivity of M components to myelin would be an attractive hypothesis in the demyelinating neuropathy seen in osteosclerotic myeloma, but has been shown infrequently.17
Recent evidence implicates a role for cytokines in some aspects of POEMS syndrome. Bone destruction in myeloma is contributed to by cytokines such as tumour necrosis factor β (TNF-β) and interleukin 1b (IL-1b), which act as osteoclastic activating factors.24 25 Increased concentrations of TNF-α have been found in the serum of patients with POEMS and early weight loss15 and have been proposed as a possible cause of the pronounced wasting. Increased concentrations of IL-1 and IL-6 have also been found in POEMS syndrome15 and in Castleman’s disease increased serum concentrations and production of IL-6 by lymph node cells have been found.26 Several workers have proposed an angioproliferative factor to account for the proliferative changes described in small blood vessels.17 27 These factors may also play a part in the neuropathy as TNF-α and IL-1 have been implicated in increased vascular permeability and TNF-α has been shown to be effective in blood-nerve barrier breakdown.28Compromise of the blood-nerve barrier is clearly a prerequisite for circulating immunoglobulin to react with myelinated nerve fibres.
TREATMENT
The neuropathy complicating multiple myeloma does not usually respond to therapy with either plasmapheresis, cytotoxic drugs, or a combination of these3 9 although there are occasional reports of benefit.29 Some cases associated with solitary plasmacytoma have improved after radiotherapy.16 30
Kyle and Dyck9 recommend radiotherapy (40–50 cGy) for patients with single or multiple osteosclerotic lesions in a limited area and have found substantial improvement of the neuropathy in more than 50%. The improvement may be slow. In patients with widespread lesions chemotherapy is advised (melphalan and prednisone).9 The combination of prednisone and cyclophosphamide has also been documented to benefit the neuropathy in some patients with POEMS syndrome.13 17 31
Neurological complications of paraproteinaemia
Monoclonal components may be detected in patients with conditions other than multiple myeloma or plasma cell dyscrasias. They may occur in lymphoid and non-lymphoid neoplasia and various autoimmune conditions. Moreover, they may be seen in the absence of any recognisable disease. These cases are classified as MGUS. The second phrase recognises the need for long term follow up of these patients. Such benign paraproteins occur in about 3% of the population but the incidence increases with age.
Peripheral neuropathy associated with MGUS
Several early case reports described an association between neuropathy and IgM paraproteins. Kahn et al 32first demonstrated a clear statistical relation between neuropathy and the presence of paraproteins. They studied 14000 serum samples from patients referred to a neurological centre. Fifty six patients had MGUS and 16 of these had neuropathy. The report by Latov et al 33 which showed binding of IgM to the myelin sheath in a patient with a demyelinating neuropathy stimulated intense interest and was confirmed by other workers.34-37Although IgG paraproteins are the most common, neuropathy is more often seen in patients with IgM paraproteinaemia. In a series of 7004 patients with MGUS from the Mayo Clinic 74% had IgG, 15% IgM, and 11% IgA paraproteins. In this group 65 patients had neuropathy; 48% of those patients had IgM paraprotein, 37% IgG, and 15% IgA.38 Among 62 patients with neuropathy and MGUS reported by Yeung et al,39 the paraprotein class was IgM in 46, IgG in 11, and IgA in five. Kelly et al 40 found that 10% of 279 patients with neuropathy without underlying systemic disease (alcohol, diabetes, etc) had an M protein on serum electrophoresis. Six per cent had MGUS, 2.5% primary amyloid, 1.1% myeloma, and there was one patient each with Waldenstrom’s macroglobulinaemia and heavy chain disease.
Neuropathy associated with IgM paraproteinaemia
CLINICAL FEATURES
The neuropathy usually presents as a slowly progressive distal sensorimotor neuropathy, predominantly affecting men36 38 39 in the sixth or seventh decade. Very occasionally the course is relapsing and remitting and the symptoms and signs purely sensory. Multifocal neuropathy has rarely been found.39 Sensory symptoms predominate at onset and ataxic and upper limb postural tremor are prominent features.36 38 39
LABORATORY INVESTIGATIONS
The CSF protein is often raised and motor conduction velocity reduced to levels within the demyelinating range.36 A characteristic finding is that of considerable prolongation of distal motor latencies representing a large distal accentuation of conduction slowing.41
Pathological changes in peripheral nerve are characterised by demyelination and fibre loss. Fibre loss is often considerable. Inflammatory infiltrate as usually lacking but hypertrophic changes are occasionally seen.37 42
Electron microscopy has shown in many cases widened myelin lamellae between major dense lines, most pronounced in the outer myelin lamellae (fig 3).36 37 42 The evidence suggests that this change results from the deposition of the paraprotein, and it is particularly pronounced in perinodal regions and Schmidt Lantermann incisures, where the antigenic target, myelin associated glycoprotein, is located.43 An interesting finding is that on teased fibre preparations tomacula (focal areas of hypermyelination) may be seen,44 a finding characteristic of hereditary sensitivity to pressure palsy, due to a deletion within the gene for PMP-22.

{kind=link}
{kind=link}
{kind=link}
Electron micrograph of a sural nerve biopsy specimen from a patient neuropathy associated with IgM paraprotein. Note that outer myelin lamellae (wl) are widened. Bar = 1 μm.
IMMUNOPATHOLOGY: PARAPROTEIN SPECIFICITY
IgM binding to surviving myelin sheaths has been shown by both direct and indirect immunofluorescent techniques.22 45Latov et al first showed that the antigenic target of some IgM paraproteins was myelin associated glycoprotein ,46 a finding confirmed by others.37 47 48 These antibodies have a similar specificity to the murine monoclonal antibody HNK-1 which recognises a carbohydrate epitope on other glycoprotein and glycolipid molecules within nerve49: PO glycoprotein,50 PMP22 glycoprotein, the glycolipids sulphated glucuronic acid paragloboside (SGPG), sulphated glucuronic acid and lactosaminyl paragloboside (SGLPG),49 and several cell adhesion molecules, N-CAM, LI, and JI glycoprotein.51However the patients’ IgM antibodies have a higher affinity for myelin associated glycoprotein than the other glycoproteins52 and react poorly with several of the other molecules. The use of specific antihuman IgM antibodies to detect the IgM paraproteins deposited in vivo has shown these mainly in the periaxonal and the outer rim of the myelin sheath, Schmidt-Lantermann incisures, and paranodal loops—that is, the region of myelin associated glycoprotein localisation. About 50% of IgM paraproteins associated with sensorimotor neuropathy react with myelin associated glycoprotein.53
IgM paraproteins may also react with GM-I gangliosides. Monoclonal or polyclonal IgM anti-GM-1 antibodies have been reported in association with a motor neuropathy with conduction block and motor neuron disease.54-56 Most of these antibodies show specificity for the terminal Gal (β1–3) Gal Nac structure.
Other antibodies show specificity for the disialosyl groups on gangliosides GD1b, GT1b, GQ1b, and others. Patients with these antibodies present with a progressive ataxic neuropathy with involvement of large sensory fibres.57 Patients with chronic sensory neuropathy have also been described with paraproteins reacting to chondroitin sulphate58 and sulphatide.56 The pathological changes in these cases is axonal degeneration.
THE ROLE OF ANTIMYELIN ASSOCIATED GLYCOPROTEIN(MAG) ANTIBODIES
The mechanism by which these antibodies cause demyelination may not be clearly understood until the role of myelin associated glycoprotein is more clearly defined. However there is accumulating evidence that the antibodies are pathogenic. As described above IgM can be demonstrated in situ in regions of myelin associated glycoprotein localisation. Focal demyelination was produced by intraneural injection of antibody into the sciatic nerve of the appropriate species.59 Tatum60 has produced the electrophysiological and pathological features of the disease by passive transfer studies into newborn chicks, which have a defective blood-nerve barrier. The question of how the antibodies cross the blood-nerve barrier in humans remains unanswered, but the electrophysiological findings of Kaku et al 41of distal slowing, suggest that the antibody leaks into nerve preferentially from the region of the neuromuscular junction where the blood nerve barrier is known to be deficient. There is therefore mounting evidence that the neuropathy associated with anti-MAG antibody can be regarded as an autoimmune disorder of nerve.
NEUROPATHY ASSOCIATED WITH IgG AND IgA PARAPROTEINAEMIA
Although IgG paraproteins are more common in the community then IgM, neuropathy is more commonly associated with the IgM.22 39 45 The course of neuropathy associated with IgG paraproteins may be chronic progressive or relapsing and remitting. Five cases of IgG paraproteinaemia and neuropathy were described by Bleasel et al 62; they followed a relapsing and remitting course and were in all respects, apart from the monoclonal protein, similar to cases of chronic inflammatory demyelinating polyneuropathy (CIDP). Although binding of IgG or IgG light chain fragments to the myelin sheath has been demonstrated in a few cases22 61 evidence for reactivity to defined myelin antigens is lacking. Three cases with sensorimotor axonal neuropathies have been described with IgG antibody reactive to neurofilament determinants.62
Relatively few cases of neuropathy associated with IgA paraproteinaemia have been described. In the series of Gosselin et al 38 there were 10 patients in whom the clinical features were similar to cases of IgG and IgM paraproteinaemia neuropathy. Three separate patients have been described in whom IgA paraproteinaemia was associated with a motor neuron-like disease.22 63 64 Hays et al 65reported a similar patient in whom the IgA paraprotein reacted to a neurofilament antigen.
TREATMENT
There is general agreement that patients with neuropathy associated with IgG and IgA paraproteins respond to therapy—either plasma exchange, intravenous immunoglobulin, or immune suppression (steroids with or without azathioprine or cyclophosphamide).9 39 61 The efficacy of treatment in patients with IgM associated neuropathy remains controversial. Benefit has been reported in several uncontrolled studies29 33 63 but not in a controlled trial.9 66 Other centres with considerable experience confirm the disappointing results of treatment in IgM paraprotein associated neuropathy.39 67 These patients fortunately progress very slowly and many may be managed conservatively without the introduction of potentially dangerous therapies (corticosteroids or immune suppression). In patients who progressively deteriorate it is our practise to try plasma exchange or intravenous immunoglobulin and introduce immunosuppressive agents on a trial basis (pulse cyclophosphamide or melphalan) only when the first treatment has failed and if patients are becoming seriously incapacitated.
Patients in whom monoclonal or polyclonal antibodies against GM-1 ganglioside are associated with a clinical syndrome of multifocal motor neuropathy with conduction block may respond to therapy with either intravenous immunoglobulin or cyclophosphamide.68
Waldenstrom’s macroglobulinaemia
Macroglobulinaemia results from an uncontrolled proliferation of lymphocytes and plasma cells which produce excessive circulating monoclonal IgM protein; the bone marrow is extensively infiltrated with lymphocytes and plasma cells. Other causes of macroglobulinaemia include IgM monoclonal gammopathy of undetermined significance, lymphoma, chronic lymphatic leukaemia, and amyloid and IgM myeloma. The clinical features usually include weakness, fatigue, and bleeding often of the oronasal region. Impaired vision, mental confusion, neuropathy and other neurological symptoms may occur. Dyspnoea and congestive cardiac failure may be evident and enlargement of spleen, liver, and lymph nodes.
STROKE AND SUBARACHNOID HAEMORRHAGE
Patients may present with sudden fatal cerebral haemorrhage or with focal brain syndromes, as a result of the bleeding tendency or from cerebral ischaemia.69 The M protein interferes with the clotting cascade, but a more important mechanism seems to be an abnormality of the platelet plug and its formation.70
ENCEPHALOPATHY: HYPERVISCOSITY SYNDROME
Bing and Neil71 first described the association of diffuse CNS disease and hyperglobulinaemia. Numerous CNS manifestations have been described in macroglobulinaemia including pyramidal tract dysfunction, dizziness, headache, ataxia, tremors, hearing loss, lethargy, organic psychosis, and coma.69 Many of these cases showed retinal change including haemorrhages, venous engorgement, papillitis, and exudates.72 Pathological studies showed lymphocyte and plasma cell infiltration of Virchow Robin spaces,73 multiple small haemorrhages within the brain, and plasma (IgM) exudation into brain parenchyma and perivascular spaces.70 Because of these findings it has been proposed that these symptoms of diffuse CNS involvement result from increased viscosity and altered vascular permeability.
The hyperviscosity syndrome is characterised by bleeding, ocular, neurological, and, rarely, cardiovascular manifestations.70 Ocular symptoms include diplopia, blurred vision, and visual failure. The retinal veins are distended and tortuous and may assume the form of a string of sausages. Haemorrhages, exudates, and papilloedema may occur. Symptoms of CNS involvement include headache, dizziness, deafness, unsteadiness, and vertigo, and impairment of consciousness which may progress to coma or death. The importance of recognising this syndrome is that treatment of the hyperviscosity by plasmapheresis can relieve most symptoms.70
MYELOPATHY
Spinal cord syndromes (spastic paraparesis or tetraparesis) may result from the hyperviscosity changes described above, in addition to the more common cerebral symptoms and signs. The spinal cord may be compromised by bony compression or cellular infiltration. Patients with macroglobulinaemia and lower motor neuron syndromes have been described and in one case signs and symptoms of primary muscular atrophy reversed after treatment with chlorambucil.74
PERIPHERAL NEUROPATHY
Sensorimotor peripheral neuropathy occurs commonly in macroglobulinaemia.75 76 The symptoms, signs, and laboratory findings are essentially the same as those already discussed for IgM paraprotein associated neuropathies.
TREATMENT
Plasmapheresis is indicated for the symptoms of the hyperviscosity syndrome. It has been reported in uncontrolled studies to improve patients with neuropathy29 77 but other reports are unfavourable.75 Occasional patients have reportedly benefited from chemotherapy alone78 and also from a combination of chemotherapy (chlorambucil) and plasmapheresis.77 There have been no controlled trials.
Cryoglobulinaemia
Cryoglobulins are serum immunoglobulins which precipitate when cooled and redissolve when heated. The temperature at which they precipitate varies but this variable rather than the amount of protein present, determines clinical expression of disease.79 They have been classified as type 1 (a single monoclonal protein, IgM, IgG, or IgA), type 2 (a mixture of monoclonal and polyclonal immunoglobulins), and type III (polyclonal immunoglobulins only). Cryoglobulins may occur without evidence of underlying disease (essential cryoglobulinaemia) or be secondary to a lymphoproliferative disorder or a chronic inflammatory or infective process. A high incidence of antibodies to hepatitis C virus has been reported in patients with essential mixed cryoglobulinaemia.80
The clinical symptoms relate to cold sensitivity and include Raynaud’s phenomenon, cyanosis or skin ulceration; skin rashes especially purpura, bleeding from mucous membranes, arthralgia, glomerulonephritis, retinal haemorrhage, and neurological complications.
COMPLICATIONS OF THE CNS
Transient ischaemic attacks manifested as transient altered consciousness and coma, blindness, hemiplegia, and seizures have been described.81 82 In such cases stenosis and occlusion of cerebral vessels without atheroma have been shown, or changes consistent with CNS vasculitis. Cerebral infarction has also been described83 84 and thrombi of hyaline acidic material within blood vessels with, perivascular haemorrhage or multiple thrombotic occlusions.84
PERIPHERAL NEUROPATHY
Peripheral neuropathy usually presents in the setting of the typical manifestations of cryoglobulinaemia, Reynaud’s phenomenon, cyanosis, skin ulceration, and neuropathic symptoms are often precipitated by cold weather.85 86 Neuropathy has been reported in up to 50% of cases.87 88 It most commonly presents as a symmetric mainly sensory neuropathy with prominent pain and hypaesthesia. Occasionally a multifocal neuropathy may occur and this may be superimposed on a symmetric sensory neuropathy. Cranial nerves may occasionally be affected and although slowly progressive some patients show a seasonal remitting and relapsing course.91 Electrophysiological studies show impaired sensory conduction and mild slowing of conduction velocity.90
Neuropathological studies show mostly axonal degeneration, or axonal degeneration with some segmental demyelination in teased fibre preparations.86 Cryoprecipitation may be seen within endoneurial blood vessels and within the vaso nervorum86and endoneurial and perineurial vasculitis have been described.90 Hence the neurological symptoms and signs in cryoglobulinaemia probably have an ischaemic basis, due either to immune complex mediated vasculitis, or the precipitation of cryoglobulins within vessels.
TREATMENT
Plasmapheresis is indicated when severe manifestations of cryoglobulinaemia are present, particularly in the early stages. Sustained clinical improvement and reduction of cryoglobulin concentrations are the end points of therapy.10Improvement of both encephalopathy and neuropathy has been described after treatment with corticosteroids; combined therapy may be indicated if vasculitis is suspected. Cold exposure should be minimised.91 Treatment with interferon-α may be beneficial in patients with serological evidence of hepatitis C infection.80
Malignant lymphoma
Malignant lymphoma is a term used to describe a heterogenous group of diseases characterised by a malignant proliferation of cells of the lymphoid system. Traditionally, two separate disorders are recognised—Hodgkin’s disease and the more common non-Hodgkin’s lymphoma. Hodgkin’s disease typically affects lymph nodal tissue, and is characterised by the presence of large, often multinucleated cells—the so called Reed-Sternberg cell—which is the hallmark of this disease. The disease can affect the liver and spleen, and, rarely, other organs such as the skin, the gastrointestinal tract, and the nervous system. In general, Hodgkin’s disease represents one of the outstanding success stories of modern cancer therapy, and even patients with advanced stage disease can now expect an 80% chance of 10 year survival.
On the other hand, non-Hodgkin’s lymphoma is a very heterogenous disease with a wide variety of clinical presentations and very varied clinical outcomes. Although lymph nodal presentation is most common, patients can often present with extranodal involvement or, more rarely, with leukaemic manifestations and marrow failure. Patients may have a very indolent course requiring little therapy over periods in excess of 20 years, or have a very aggressive course with short survival. Currently non-Hodgkin’s lymphoma is going through yet another classification debate, epitomised by proponents of the revised European American lymphoma (REAL) classification.
Neurological complications may occur when the disease is active or seems to be in remission. They result from infiltration of the meninges or neural parenchyme, compression of spinal cord or nerve roots, infections of the CNS, or as complications of treatment. Neurological complications are more common when leukaemic conversion has occurred and are often seen in Burkitt’s lymphoma.
LEPTOMENINGEAL LYMPHOMA
Involvement of the meninges usually occurs in non-Hodgkin’s lymphoma of diffuse rather than nodular histology92 and with undifferentiated cell type.93 It is more often seen when the bone marrow or peripheral blood is involved and in younger patients.94 Tumour cells may invade the meninges by haematogenous spread or by infiltration along perforating blood vessels from the medullary bone marrow.95 Tumour masses involving the dura may invade the subarachnoid space and be disseminated throughout the spinal fluid pathway.96 The meninges of the base of brain and spinal cord are often involved.97
The symptoms and signs include those of increased intracranial pressure and meningitis affecting the base of brain and spinal roots; headache, vomiting, irritability, confusion, papilloedema, seizures, and cranial nerve and spinal radicular involvement. Cranial nerves VII, VI, and III are most commonly affected. Involvement of spinal roots may cause pain, paraesthesiae, or weakness, most often at the lumbar or sacral level and sphincter dysfunction and impotence result.96
Examination of the spinal fluid is mandatory in making the diagnosis, in particular the demonstration of malignant cells in the CSF. Raised CSF protein may be found and the glucose may be decreased, but both may be unaffected.95 The demonstration of meningeal enhancement or of enhancing nodules on nerve roots by MRI, provides suggestive evidence of meningeal lymphoma, particularly if infective meningitis can be excluded.
Steroids, intrathecal chemotherapy, and focal irradiation may each play a part in therapy. Methotrexate or cytosine arabinoside are usually given intraventricularly by means of a CSF reservoir. Radiotherapy may be directed to clinically or radiographically demonstrated focal lesions and a combination of radiotherapy and intrathecal chemotherapy has been shown to improve survival.92
EPIDURAL LYMPHOMA
Intracranial, epidural, and subdural lymphoma occur in less than 10% of cases of Hodgkin’s disease and non-Hodgkin’s lymphoma.98 These lesions may be located over the hemispheres but are commonly found at the skull base. Epidural masses may cause focal cerebral lesions, which will depend on their site (hemiparesis, dysphasia, altered cognition, or seizures). There may be headache and other signs of increased intracranial pressure. Basal lymphoma may compress cranial nerves or involve the pituitary and hypothalamus. Subdural lymphoma may present as subdural haematoma as it may be accompanied by fluid effusion.99
Spinal cord involvement by lymphoma derives either from tumour which invades the epidural space from paravertebral lymph nodes through intervertebral foramina, or extension from lymphomatous vertebral bodies. Compression of the spinal cord by the tumour mass is usually accompanied by symptoms and signs of nerve root compression at this level. Occasional intramedullary invasion may result from tumour growth along nerve roots. The symptoms and signs are those of back and nerve root pain followed by the features of spinal cord compression: weakness, sensory, and sphincter disturbances. Early diagnosis and treatment are essential to prevent permanent disability, and to provide the optimum chance for recovery. MRI and CSF examination are indicated to confirm the diagnosis and localise the area of involvement; therapy consists of high dose steroids when this complication is suspected followed by radiotherapy on radiographic confirmation. Surgical decompression is rarely indicated except when the diagnosis is uncertain or other complications—for example, epidural abscess—are suspected.
INTRACRANIAL LESIONS
Mass lesions intracranially are more common in patients with non-Hodgkin’s lymphoma but the overall incidence is low.96 98 They mostly derive from meningeal deposits but haematogenous metastases do occur.
The symptoms and signs depend on the site of the lesion and the diagnosis should be confirmed by CT and MRI and sometimes stereotactic biopsy. These lesions are often responsive to steroids and radiotherapy but if the meninges are involved intrathecal chemotherapy may be indicated.
PERIPHERAL NEUROPATHY
Clinical evidence of neuropathy is uncommon (8%) but when electrophysiological studies are used a higher incidence (35%) may be shown.7
Several different clinical types of neuropathy are recognised.
SENSORIMOTOR NEUROPATHY
Acute polyneuropathy of the Guillain-Barré type occurs in patients with lymphoma, more commonly in patients with Hodgkin’s disease.100 Lisak et al 100 found abnormal immune responses in their patients and suggested that immune suppression predisposed to the occurrence of the neuropathy. The pathological changes are typical of Guillain-Barré syndrome. Subacute neuropathy has been described mainly in non-Hodgkin’s lymphoma and is usually accompanied by lymphomatous infiltration of peripheral nerves and nerve roots.
Relapsing and remitting neuropathy is rare in patients with lymphoma. Pathological studies in these cases have shown macrophage mediated demyelination and axonal loss. Sensorimotor neuropathy associated with lymphoma usually follows a chronic progressive course. It may precede or follow the diagnosis of lymphoma.101 Axonal degeneration and segmental demyelination have been reported, with occasional evidence of microvasculitis.102
SUBACUTE MOTOR NEUROPATHY
A subacute asymmetric lower motor neuron syndrome, affecting primarily the lower limbs, has been described. It may spontaneously remit. Pathological studies showed anterior horn cell degeneration and motor root demyelination103 but no tumour infiltration or inflammation.
SENSORY NEUROPATHY
Few cases of sensory neuropathy complicating lymphoma have been described. The pathological changes are similar to those described in sensory neuropathy complicating carcinoma.
Because mediastinal lymph nodes are often involved in Hodgkin’s disease the recurrent laryngeal nerve, phrenic nerve, and sympathetic chain may be compromised by local tumour masses.
LYMPHOMA ASSOCIATED WITH HIV
The neurological manifestations of HIV/AIDS infection are protean, and can involve any component of the nervous system.104The disorders include HIV dementia, CNS infection and neoplasms, vascular complications, peripheral neuropathies, and myopathies.
The primary CNS lymphoma associated with HIV/AIDS infection has attracted considerable attention105 not only because of its increasing incidence but also because of its interesting biology. It seems that all cases of primary lymphoma associated with HIV have integration of the EBV genome into the malignant cells,106raising the prospect of an aetiological role for the EBV virus. Patients often present with memory loss, confusion, and lethargy, but may also present with epilepsy. Radiology shows one or more contrast enhancing lesions which may be difficult to differentiate from toxoplasmosis. In practical terms this disease continues to have a poor prognosis.107
Bone marrow transplantation
Neurological complications occur in about 70% of patients with allogeneic bone marrow transplantation.108
The immunosuppressive regime which usually consists of cyclophosphamide and low dose total body irradiation rarely produces neurological complications. However, because of the underlying disease process and myeloablative therapy, patients undergoing bone marrow transplantation are usually severely immunosuppressed and hence prone to infection. Within the first month, when patients are granulocytopenic, bacterial, viral, or fungal infections may occur. Despite engraftment patients remain immunologically compromised for up to one year and are prone to viral (particularly cytomegalovirus) and protozoan infection (especially toxoplasmosis).109
A major complication after allogeneic bone marrow transplantation is graft versus host disease (GVHD), in which immunologically competent donor lymphocytes attack host tissue. Chronic rather than acute GVHD is associated with neurological complications. Chronic neuromuscular disorders are prominent among these. Polymyositis indistinguishable from idiopathic polymyositis is well described.110 111 It responds to treatment directed to the GVHD—that is, immunosuppression.
Myasthenia gravis, accompanied by raised concentrations of acetylcholine receptor antibody and responsive to anticholinesterase and immunosuppressive agents is also well described.112
Peripheral neuropathy has also been reported. The features described are those of CIDP,113 114 and most cases have responded to immunosuppressive agents.
Although there have been reports in which the patients with GVHD have shown lymphocytic infiltration within the brain no recognised pattern of disease has emerged for CNS involvement.115 An interesting animal model of GVHD has been described in which massive lymphocytic infiltration occurs within the brain accompanied by pronounced upregulation of MHC class I and II molecules.116 No human equivalent of this condition has been described.
Cerebral infarction resulting from infective endocarditis or non-bacterial thrombotic endocarditis has been reported in about 10% of necropsy series of patients with BMT. The reasons for this complication are unknown.117
Neurological complications of treatment
It is important to remember that neurological signs and symptoms in patients with haematological disorders may result from infection or complications of therapy.
Radiation therapy
Radiation therapy which is so effective in the management of lymphoma, unfortunately has a wide variety of neurological adverse reactions. The brain, spinal cord, or peripheral nerve may be affected. Tissue damage is to some degree dose dependent but there is also an idiosyncratic element. Concomitant chemotherapy and systemic illness may increase the susceptibility to radiation damage.94 The combination of cranial irradiation with high dose intravenous or intrathecal methotrexate is particularly associated with an increased occurrence of encephalopathy, which may take the form of a disseminated necrotising leukoencephalopathy. This is seldom seen with doses of irradiation <2000 cGY.118
ENCEPHALOPATHY
Encephalopathy is more likely to develop if methotrexate is given during or after cranial irradiation and it has been proposed that the second damages the blood-brain barrier allowing methotrexate more ready access to the brain parenchyma.118 Acute, subacute, or chronic encephalopathy, myelopathy, or peripheral neuropathy may occur. Acute encephalopathy usually occurs within the first week of therapy and produces lethargy, fever, headache, nausea, and vomiting. It responds to corticosteroid therapy and may be due to radiation induced cerebral oedema. A subacute encephalopathy occurring after several weeks is much less common and presents with headache, drowsiness, seizures, nausea, vertigo and ataxia, and brain stem or cerebellar features. It usually improves spontaneously but may be aided by corticosteroid therapy. White matter changes are evident on MRI or CT. Delayed or chronic encephalopathy is more common and may present with focal defects, progressive dementia, apraxia, or seizures. PET is helpful in differentiating this condition from intracranial malignancy. The pathological changes include mineralising microangiopathy, disseminated necrotising. leukoencephalopathy, calcification, axonal degeneration, and demyelination.94 96
MYELOPATHY
An early transient myelopathy occurring six to 12 weeks after treatment is uncommon and usually recovers within three to 12 months. Delayed progressive myelopathy is more common and may occur from six months to six years after radiotherapy. Sensory, motor, and sphincteric symptoms and signs develop related to the site of involvement. The incidence may be as high as 15% in patients with Hodgkin’s disease.119 The course is usually inexorably progressive.
PERIPHERAL NEUROPATHY
Peripheral nerves are relatively resistant to ionising radiation. The brachial plexus, however, may be involved after irradiation of the axilla or cervical nodes. The clinical features are of painless progressive sensorimotor changes within the distribution of the plexus. These changes usually occur from six months to 15 years after irradiation and are usually very slowly progressive. Pathological changes are characterised by dense epineurial fibrosis, with demyelination and axonal loss. Blood vessels within the area are thickened and hyalinised with lumen reduction or occlusion.
Infection
Patients with lymphoma have an increased susceptibility to infection for several reasons; disease associated impaired immunity, immunosuppressive therapy, splenectomy, and the insertion of CSF reservoirs and shunts.
BACTERIAL INFECTIONS
There is a well documented association between lymphoma andListeria monocytogenes.120 It usually presents as an acute meningitis but may be accompanied by brain stem signs. Gram negative organisms, E coli, Pseudomonas, Proteus,and Pneumococcus may also be responsible.121 122
FUNGAL INFECTIONS
Fungal and bacterial infections account for about 40% of CNS infections in lymphoma. Candida, aspergillus, and cryptococcus are the most frequent fungi involved.
VIRAL INFECTIONS
Herpes zoster is the most common viral infection in patients with lymphoma, affecting about 20% of patients with Hodgkin’s disease at some time during its course.123 It may present as meningitis, encephalitis, myelitis, peripheral neuritis particularly affecting the thoracic root zones, and ophthalmic zoster. The symptoms and signs are similar to those of zoster in the general population except that generalised infection is seen more commonly in patients with lymphoma.97
Infection with cytomegalovirus, and toxoplasmosis have been reported.124 The first may produce encephalitis and retinitis and the second encephalitis or focal signs due to mass lesions.
Progressive multifocal leukoencephalopathy has often been associated with lymphoma,125 and was first described by Aströmet al 126 in patients with chronic lymphatic leukaemia and lymphoma.
Toxic effects of chemotherapy
ENCEPHALOPATHY
Acute encephalopathy may occur with several antineoplastic agents used in the treatment of lymphoma and other bone marrow malignancies. It is a common side effect of L-asparaginase. It may be seen with high dose intravenous or intrathecal cytosine arabinoside and methotrexate. Methotrexate therapy may result in acute, subacute, or chronic encephalopathy. Acute encephalopathy results in confusion, lethargy, and seizures within 24 hours of treatment and usually recovers fully. BrainMRI usually shows no change. Subacute encephalopathy presents similarly except that focal signs may be evident and it occurs days to weeks after treatment.117MRI usually shows increased signal intensity in the periventricular regions of the white matter on T2 weighted images and recovery usually occurs spontaneously over a few days. Encephalopathy after chronic high dose methotrexate or intrathecal therapy is usually of gradual onset and neurological deficits persist. This complication limits the dose or further treatment. Encephalopathy after vincristine therapy is rare. SIADH is another rare complication.127
CEREBELLAR DYSFUNCTION
Acute and subacute cerebellar dysfunction (and encephalopathy) may follow high dose intravenous cytosine arabinoside. Presenting features include nystagmus and truncal ataxia followed by dysarthria, confusion and lethargy. Symptoms usually resolve after stopping the drug. Loss of Pürkinje cells may be evident on histological sections of the cerebellum.128 A similar syndrome may be seen with 5-fluorouracil.
MYELOPATHY
Transient and permanent spinal cord damage have been reported after intrathecal administration of methotrexate and cytosine arabinoside but this is a rare complication.
PERIPHERAL NEUROPATHY
Peripheral neuropathy is well known as a complication of vinca alkaloid therapy and its occurrence is dose related. It is the dose limiting factor in the use of this drug. Sensory abnormalities may occur but weakness which develops rapidly is the major manifestation of the neuropathy. The pathological findings are those of axonal degeneration.129 Vinca alkaloids bind to tubulin, and the antimitotic effect results from their action on spindle microtubules in dividing cells. In nerve they cause microtubular breakdown and hence reduce axonal transport and result in a “dying back” neuropathy.
Occasional cases of subacute neuropathy after high dose intravenous methotrexate have been described. This neuropathy seems to resolve over a few days.
Summary
Neurological complications are commonly encountered in disorders of the bone marrow and they result from various pathological processes. Compression of nervous tissue, brain, spinal cord, or nerve may occur from tumour masses—lymphoma, myeloma, or after bone destruction as in myeloma. Neurological dysfunction may result indirectly from aberrant immune responses. Impaired immunity, which commonly accompanies bone marrow disease permits the development of opportunistic infection within the nervous system of which herpes zoster radiculitis and progressive multifocal leukoencephalopathy are two well recognised examples in patients with lymphoma. On the other hand, autoantibodies produced by a dysregulated bone marrow are responsible for the neuropathy seen in association with paraproteinemia, myeloma, and macroglobulinaemia. Excess immunoglobulin may result in hyperviscosity causing multifocal and diffuse central nervous system symptoms in patients with macroglobulinaemia. Cryoglobulins may cause ischaemic symptoms and signs in the central and peripheral nervous systems due to precipitation within vessels or because of immune complex mediated vasculitis. An increased production of cytokines in addition to immunoglobulins contribute to the neurological features associated with POEMS syndrome. In “graft versus host” disease after bone marrow transplantation, immunologically competent donor lymphocytes attack host tissue, producing chronic neuromuscular disorders including polymyositis, myasthenia gravis, and chronic inflammatory demyelinating polyradiculoneuropathy. The treatment of bone marrow disorders may itself be toxic to the nervous system; radiation myelitis or neuritis and vincristine neuropathy are examples commonly encountered by the neurologist. These treatments may also impair a depressed immune system further favouring opportunistic nervous system infection. Understanding the mechanism of neurological symptom production in bone marrow disorders has in some instances permitted the development of rational therapy.
Acknowledgments
We thank the National Health and Medical Research Council of Australia and the National Multiple Sclerosis Society of Australia.